Continuing Education Activity
Castleman disease is a lymphoproliferative disorder with a nondefinite etiology and can be classified into subtypes based on clinical and histological manifestations. Therefore, this condition's high morbidity and complications can be avoided with early diagnosis and prompt treatment. This activity explains the evaluation and treatment of Castleman disease and highlights the role of the interprofessional team in evaluating and treating patients with this condition.
Objectives:
- Describe the pathophysiology of Castleman disease.
- Outline the clinical signs and symptoms, common physical and laboratory findings in Castleman disease.
- Review the epidemiology and commonly associated diseases with Castleman disease.
- Summarize the management and treatment options available for Castleman disease.
Introduction
Castleman disease (CD) is a rare, nonclonal lymphoproliferative disorder having distinct subtypes depending on its etiology, pathology, and clinical presentation. It can affect lymph nodes of any body region, imitating both benign and malignant malformations, including the neck, chest, abdomen, and pelvis. Pathologically it can be classified as hyaline vascular type (HV-CD), plasma cell type, mixed type, and human herpesvirus (HHV)-8 associated Castleman disease.[1]
Clinically it manifests as the more common unicentric ( localized or unifocal ) Castleman disease (UCD) and the less common multicentric ( generalized or multifocal ) Castleman disease (MCD).[2]
Etiology
There is very little knowledge available about the etiology of this disorder, but some evidence that has been collected claims it to be a result of impaired immunoregulation that causes abundant proliferation of B lymphocytes and plasma cells in lymphoid organs. These conditions can result from chronic low-grade inflammation, lymphoid-hamartomatous hyperplasia, viral infections, abnormal modulation of cytokines, and angiogenesis.[3]
In addition, a very close association of the disease with HIV has been discovered, which puts immunodeficiency on the list as well. Particularly in MCD, the causative factors include human herpesvirus (HHV)-8, which causes deregulation of the inflammatory mediators like CD20 and interleukin (IL)-6.[4]
Almost all cases of HIV-associated Castleman disease are HHV-8 positive, compared to 40 to 50% of non-HIV CD. The remaining cases unrelated to HIV or HHV-8, termed idiopathic multicentric CD (iMCD), have no established etiology to date.
Epidemiology
The lack of proper knowledge and even a formal definition of Castleman disease made it challenging to assess its epidemiology. A study was conducted in 2008 by Simpson which showed that:
- The incidence of UCD is 16 per million patient-years and affects all age groups.
- The incidence of HHV-8 associated CD varies widely, but it is more common in HIV-positive men.
- The incidence of iMCD is 5 per million patient-years.[5]
Another study conducted in the US from 2000 to 2009 concluded that out of 59 patients with MCD, 61% were males with the mean age of 53 years, and 68% were white.[6]
The hyaline-vascular variant is more common than the plasma-cell type, comprising 91% of the total cases.[4]
Its predominance in any specific sex or race is not noted. Castleman disease usually develops in most patients before reaching 30, although the age range is broad.
Pathophysiology
Pathogenetically, plasma cell Castleman disease correlates with the overproduction of IL-6 and overexpression of IL-6 receptors that lead to the proliferation of B lymphocytes, vasculogenesis, resulting in a series of systemic symptoms such as fever, anemia, hypoproteinemia, and proteinuria. In some cases of mixed CD, IL-6 increase also is found to favor hepcidin production from the liver that can cause iron-deficiency anemia.[7]
Nontypical plasma cell populations with limited light chains in Immunoglobulin G (IgG) and Immunoglobulin A (IgA) can be seen. The genome of the human lymphotropic virus HHV-8 in HHV-8 associated CD encodes a viral analog (vIL)-6 of IL-6 that further stimulates angiogenesis, hematopoietic and lymphocytic activity.[8]
Histopathology
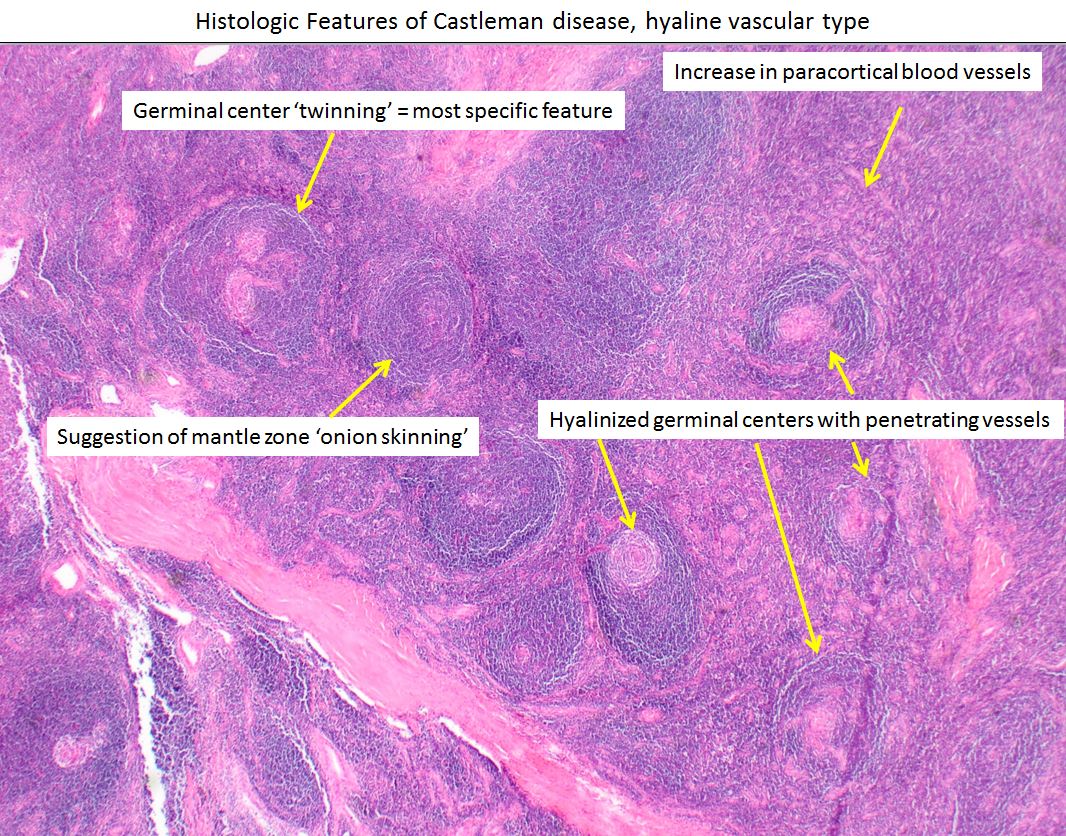
The hyaline vascular type has an increased number of lymphoid follicles with a broad mantle zone and hypoplastic germinal center. Centrally placed germinal center is surrounded with small lymphocytes arranged in concentric circles, forming an "onion skin-like" structure. The center of these follicles is penetrated by hyalinized capillaries resembling a "lollipop" (also known as the lollipop sign). Strong endothelial vascular proliferation and hyalinization in the interfollicular zones along with a varying number of small lymphocytes and immature plasma cells, and prominent follicular dendritic cells (FDCs) occasionally appearing dysplastic can be seen.[9]
A high number of plasmacytoid dendritic cells can be found around these lesions. Mature lymphocyte infiltration can be observed in plasma cell CD with maintained mantle zone and germinal zone proportions. The interfollicular zones are filled with mature plasma cells and are less vascular than HV-CD. Sometimes irregular aggregates of polyclonal plasmablasts can be seen in the mantle zone of B-lymphocytes.[9]
The mixed type has the characteristics of both HV-CD and the plasma cell type. HHV-8 associated CD manifests as significant proliferation of angiogenesis between the lymphoid follicles with unclear boundaries and immature plasmablasts that might have restricted lambda light chains in specific immunoglobulins.
History and Physical
Clinically, unicentric Castleman disease presents as a slow-growing, non-malignant painless solitary mass manifesting at a single anatomic site that is generally asymptomatic at first but starts producing symptoms when the surrounding structures are compressed due to its growing size.[10][11]
In contrast, multicentric CD occurs at multiple sites and is characterized by a proinflammatory response giving rise to the so-called B symptoms, including fever, night sweats, malaise, and weight loss. Exaggerated effects of elevated serum IL-6 levels cause various symptoms, including lymph node enlargement, hepatosplenomegaly, polyclonal hypergammaglobulinemia- owing to its growth and differentiation factors for both plasma and lymphocytes. Elevated fibrinogen levels cause deep vein thrombosis and thromboembolic disorders, high hepcidin levels cause anemia, increased VEGF levels result in angiogenesis and vascular permeability, which along with hypoalbuminemia induces edema, ascites, pleural and pericardial effusions, or even generalized anasarca.
In extreme cases of iMCD, multiple organ failure with renal insufficiency can ensue, often resulting in death.[12]
Evaluation
The diagnosis of Castleman disease is always a challenge in clinical experience, as it does not have specific features that could be distinguished from other diseases causing lymphadenopathies.[13] Therefore, the diagnosis should be finalized when the patient meets both major criteria, at least two of the minor criteria and one laboratory abnormality.
Major Criteria
- Histopathologic screening of lymph nodes is done to assess a single node involvement (suggestive of UCD) or a multimode involvement (suggesting MCD) after excluding other infectious, malignant, and autoimmune disorders that exhibit their characteristic features in the lymph nodes. In addition, their characteristic lymph node features are also noted.
- The lymph node size must be enlarged.
Minor Criteria
Laboratory
- Elevated CRP or ESR
- Anemia
- Thrombocytopenia or thrombocytosis
- Hypoalbuminemia
- Renal dysfunction or proteinuria
- Polyclonal hypergammaglobulinemia
Clinical
- B symptoms: fever, weight loss, night sweats, fatigue
- Splenomegaly or Hepatomegaly
- Fluid accumulation( edema, anasarca, pleural effusion)
IL-6 has proven to be a specific biomarker for this disease, contributing to its pathogenesis, symptomatology, and histopathology. However, raised blood levels of IL-6 and soluble IL-2 receptor (sIL2R), VEGF, IgA, IgE, lactate dehydrogenase, β-2-microglobulin do not specifically indicate the presence of this disease but are essential for supporting its diagnosis.[14]
The patient must also undergo serology tests for HIV and HHV-8 and search for HHV-8 DNA in peripheral blood by polymerase chain reactive (PCR) to discover associations with CD.
Before formulating a treatment plan, physicians should perform a thorough clinical staging to detect other sites of involvement. An evaluation including serum protein electrophoresis, bone marrow examination, CT scan of the chest, abdomen, and pelvis, radiographic skeletal survey, and gallium scan provides essential information in detecting the extent of the disease and also distinguishes multicentric from localized Castleman disease.[4]
These outlined criteria require careful collaboration between the clinical team and laboratory physicians. Definitive diagnosis should only be made when all other causes of lymphadenopathy are investigated and excluded. Elimination of other diseases may require biopsies, serologic or histologic studies, and clinical correlation.
Treatment / Management
Castleman disease is a rare disease, with minimum availability of therapeutic options. As a unicentric Castleman disease lesion is localized, complete surgical resection of the tumor is the best treatment available.
Many treatment options are available for treating multicentric CD, including surgery, cytotoxic chemotherapy with or without corticosteroids, and autologous stem cell transplantation (ASCT) with varying outcomes. Better results have been observed by targetting CD20 and IL-6 pathways and HHV-8 replication.[15]
Monoclonal antibodies interrupting the IL-6 signaling cascade, including the anti–IL-6R antibody Tocilizumab and Siltuximab, an anti–IL-6 antibody, tend to be highly effective and are considered to be the first-line therapy, especially for highly symptomatic patients without being HIV or HHV-8 positive. Antiviral therapy comprising of ganciclovir, foscarnet, and cidofovir is proven effective against HHV-8 replication pathways. Novel immunomodulatory agents like rituximab, thalidomide, bortezomib, IL-1 antagonist anakinra, and interferon-alpha have somehow shown promising results as well with minimum side effects.[14]
Chemotherapy and immunomodulatory drugs are best reserved for the relapse scenarios. However, it should be kept in mind that not all CD cases respond to therapy and get cured.
Differential Diagnosis
Castleman disease is easily confused with lymphoma or other solid tumors. Therefore, it is essential to properly diagnose the exact type of CD and distinguish it from other diseases by clinical history and laboratory diagnostic measures with additional imaging techniques for prompt treatment and management procedures. Even though CD was defined as a benign lymphoproliferative disease, the systemic forms are particularly associated with related neoplasms and autoimmune disorders like Kaposi sarcoma and Follicular dendritic cell (FDC) tumors.[4] Lymphomas, especially Hodgkin and angioimmunoblastic T-cell lymphoma, are also known for polymorphous infiltrate and hypervascularity, are well-known to mimic Castleman disease.
The hyaline vascular CD can be strongly connected to FDC tumors, including dendritic cell sarcomas and neoplasms of the stroma. In addition, IL-6 release in CD may invoke molecular mimicry and epitope spreading, initiating a lichenoid interface dermatitis, leading to autoantibody production. Finally, it can be associated with a fatal autoimmune blistering disease called paraneoplastic pemphigus in patients with treatment-resistant erosive mucosal lesions.[16]
POEMS syndrome is characterized by polyneuropathy, organomegaly, endocrinopathy, M protein, and skin changes and is a rare plasma cell disease with multisystem involvement. Around 30% of patients with POEMS disease also have multicentric CD, more precisely the HHV-8 positive variant. In addition, many findings of systemic CD-like ascites, lymphadenopathy, hepatosplenomegaly, and polyneuropathy overlap POEMS syndrome.
The HHV-8 associated CD and Kaposi sarcoma are vascular lesions caused by the same viral agent, HHV-8. Therefore, they frequently appear alongside HIV as well.
Some IgG4-related diseases like sclerosing cholangitis, Kuttner tumor, retroperitoneal fibrosis, Mikulicz disease, autoimmune pancreatitis are associated with hypertrophy of lymph nodes, similar to that in multicentric CD. However, these two diseases can be differentiated based on laboratory findings, i.e., raised serum IgG4 levels and IgG4-positive plasma infiltrates in the IgG4-related diseases and IL-6 elevation in Castleman disease.[15]
Prognosis
UCD being able to be eliminated with surgical resection has a good prognosis. However, since multiple systemic disorders involve MCD and sometimes concomitant diseases like HIV and HHV-8 can also be present, specific chemotherapy regimens have not been assigned, and better treatment methods still need to be explored. Moreover, along with the deficient treatment options, nonspecific clinical features that often go unnoticed with limited diagnostic measures also lead to delayed confirmation of its diagnosis. Its prognosis is, therefore, abysmal.[17]
Both UCD and MCD can sometimes progress to non-Hodgkin lymphoma (NHL).[12]
Complications
The clinical complications of Castleman disease are dependant on the clinical and histopathological subtypes. A study was performed analyzing the clinical manifestations in 53 patients with CD, which revealed that only 32 developed complications involving the skin, hemopoietic and internal organs. paraneoplastic pemphigus (PNP) and lung abnormalities like bronchiolitis obliterans (BO) are the two most critical rare diseases occurring with the UCD.
In contrast, hemopoietic (thromboembolic disorders and inflammatory reactions) and internal organ involvement (renal, hepatic) are mainly associated with MCD. The survival rate is significant in patients without these complications. Therefore, early diagnosis for these manifestations is necessary for tailoring appropriate therapy options, leading to a better prognosis.[18]
Deterrence and Patient Education
A doctor should examine it as soon as any lymph node swelling is noticed that persists for a longer time. People who have a history of HIV or HHV-8 should be even more careful when facing systemic abnormalities. Patients with MCD should go for regular checkups to avoid any fatal complications.
Enhancing Healthcare Team Outcomes
Castleman disease is a rare lymphoproliferative disorder, and its clinical characteristics, laboratory findings, and radiological images do not provide much specific information about it. It is a real challenge to distinguish it from other lymphadenopathies. The subtypes can also be only differentiated on the causative factors, morphologic features of lymph nodes, and pathophysiology. The symptoms like weight loss, malaise, nausea, lymph node spleen, and hepatic enlargement are vague and can be caused by any lymph node disease. It is only with histological patterns that CD can be accurately diagnosed with radiological images. A precise diagnosis can be made in a proper clinical setting when the signs and histological features can provide enough evidence for this disease.
The clinician and histopathologist must consistently communicate to collaborate on relevant findings before diagnosing the disorder as Castleman disease. While the general surgeon is almost always involved in the care of patients subjected to surgical resection of the lesion in UCD, it is essential to consult with an interprofessional team of specialists, including an anesthesiologist, oncologist, and vascular surgeon. The nurses are also vital members of the interprofessional group as they will monitor the patient's vital signs and assist with the education of the patient and family. The pharmacist will ensure that the patient is on the appropriate analgesics, antiemetics, and appropriate antibiotics for pain and wound infection in the postoperative period.
The pharmacist also provides antiviral, chemotherapy, and immunomodulatory drugs prescribed for MCD. The radiologist also plays a vital role in determining the disorder. Without a proper history, the radiologist may not know what to look for or what additional radiologic exams may be needed. This problem gets even more complex when MCD is associated with HIV and HHV-8 or other diseases. The American College of Radiology Appropriateness Criteria is evidence-based guidelines for specific clinical disorders reviewed by an interprofessional expert committee every three years. The current guidelines have been developed after an exhaustive review of current medical literature from peer-reviewed journals to determine the committee's appropriateness of radiological imaging and treatment procedures. In cases where evidence is not definitive or minimal, expert opinion from the specialist may recommend the type of imaging or treatment.
The outcomes and prognosis of CD depend on its cause and pathogenicity. However, prompt consultation with an interprofessional group of specialists is recommended to improve outcomes.