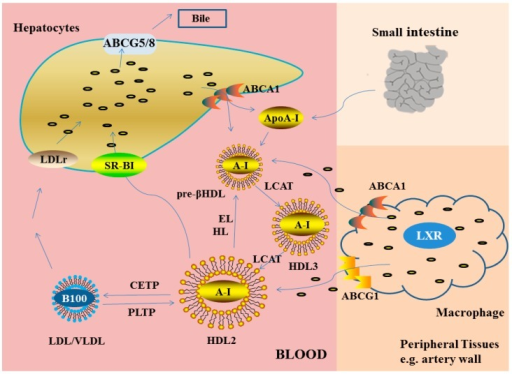
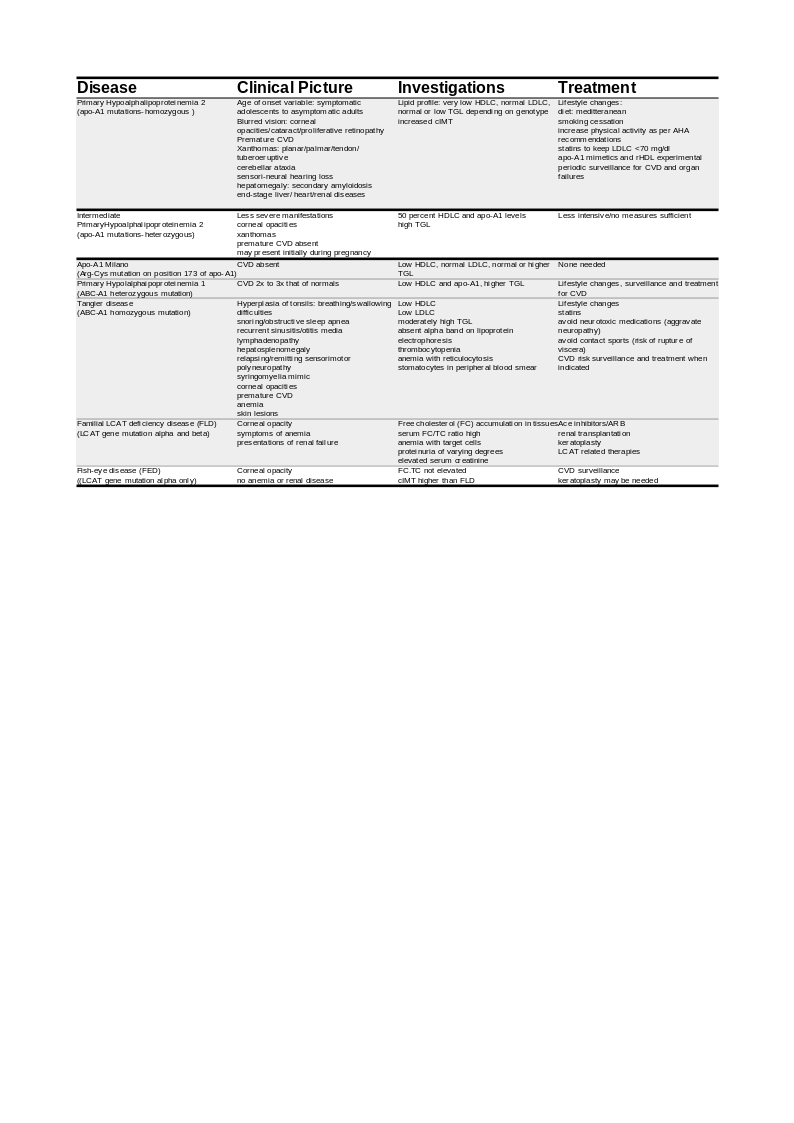
[1]
Genest J Jr, Bard JM, Fruchart JC, Ordovas JM, Schaefer EJ. Familial hypoalphalipoproteinemia in premature coronary artery disease. Arteriosclerosis and thrombosis : a journal of vascular biology. 1993 Dec:13(12):1728-37
[PubMed PMID: 8241092]
[2]
Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults. Executive Summary of The Third Report of The National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation, And Treatment of High Blood Cholesterol In Adults (Adult Treatment Panel III). JAMA. 2001 May 16:285(19):2486-97
[PubMed PMID: 11368702]
[3]
Vergani C, Bettale G. Familial hypo-alpha-lipoproteinemia. Clinica chimica acta; international journal of clinical chemistry. 1981 Jul 18:114(1):45-52
[PubMed PMID: 7249374]
[4]
Glueck CJ, Daniels SR, Bates S, Benton C, Tracy T, Third JL. Pediatric victims of unexplained stroke and their families: familial lipid and lipoprotein abnormalities. Pediatrics. 1982 Mar:69(3):308-16
[PubMed PMID: 6977760]
[5]
Cohen JC, Kiss RS, Pertsemlidis A, Marcel YL, McPherson R, Hobbs HH. Multiple rare alleles contribute to low plasma levels of HDL cholesterol. Science (New York, N.Y.). 2004 Aug 6:305(5685):869-72
[PubMed PMID: 15297675]
[6]
Brooks-Wilson A, Marcil M, Clee SM, Zhang LH, Roomp K, van Dam M, Yu L, Brewer C, Collins JA, Molhuizen HO, Loubser O, Ouelette BF, Fichter K, Ashbourne-Excoffon KJ, Sensen CW, Scherer S, Mott S, Denis M, Martindale D, Frohlich J, Morgan K, Koop B, Pimstone S, Kastelein JJ, Genest J Jr, Hayden MR. Mutations in ABC1 in Tangier disease and familial high-density lipoprotein deficiency. Nature genetics. 1999 Aug:22(4):336-45
[PubMed PMID: 10431236]
[7]
Soro A, Jauhiainen M, Ehnholm C, Taskinen MR. Determinants of low HDL levels in familial combined hyperlipidemia. Journal of lipid research. 2003 Aug:44(8):1536-44
[PubMed PMID: 12777471]
[8]
Kuivenhoven JA, Jukema JW, Zwinderman AH, de Knijff P, McPherson R, Bruschke AV, Lie KI, Kastelein JJ. The role of a common variant of the cholesteryl ester transfer protein gene in the progression of coronary atherosclerosis. The Regression Growth Evaluation Statin Study Group. The New England journal of medicine. 1998 Jan 8:338(2):86-93
[PubMed PMID: 9420339]
[9]
Nordestgaard BG, Abildgaard S, Wittrup HH, Steffensen R, Jensen G, Tybjaerg-Hansen A. Heterozygous lipoprotein lipase deficiency: frequency in the general population, effect on plasma lipid levels, and risk of ischemic heart disease. Circulation. 1997 Sep 16:96(6):1737-44
[PubMed PMID: 9323055]
[10]
Zannis VI, Chroni A, Krieger M. Role of apoA-I, ABCA1, LCAT, and SR-BI in the biogenesis of HDL. Journal of molecular medicine (Berlin, Germany). 2006 Apr:84(4):276-94
[PubMed PMID: 16501936]
[11]
van Leeuwen HJ, Heezius EC, Dallinga GM, van Strijp JA, Verhoef J, van Kessel KP. Lipoprotein metabolism in patients with severe sepsis. Critical care medicine. 2003 May:31(5):1359-66
[PubMed PMID: 12771603]
[12]
McMahon M, Grossman J, FitzGerald J, Dahlin-Lee E, Wallace DJ, Thong BY, Badsha H, Kalunian K, Charles C, Navab M, Fogelman AM, Hahn BH. Proinflammatory high-density lipoprotein as a biomarker for atherosclerosis in patients with systemic lupus erythematosus and rheumatoid arthritis. Arthritis and rheumatism. 2006 Aug:54(8):2541-9
[PubMed PMID: 16868975]
[13]
van Leuven SI, Hezemans R, Levels JH, Snoek S, Stokkers PC, Hovingh GK, Kastelein JJ, Stroes ES, de Groot E, Hommes DW. Enhanced atherogenesis and altered high density lipoprotein in patients with Crohn's disease. Journal of lipid research. 2007 Dec:48(12):2640-6
[PubMed PMID: 17890779]
[14]
Murali MR, Kratz A, Finberg KE. Case records of the Massachusetts General Hospital. Case 40-2006. A 64-year-old man with anemia and a low level of HDL cholesterol. The New England journal of medicine. 2006 Dec 28:355(26):2772-9
[PubMed PMID: 17192544]
Level 3 (low-level) evidence
[15]
Kasiske BL, Ma JZ, Kalil RS, Louis TA. Effects of antihypertensive therapy on serum lipids. Annals of internal medicine. 1995 Jan 15:122(2):133-41
[PubMed PMID: 7992988]
[16]
Wallace RB, Hunninghake DB, Reiland S, Barrett-Connor E, Mackenthun A, Hoover J, Wahl P. Alterations of plasma high-density lipoprotein cholesterol levels associated with consumption of selected medications. The Lipid Research Clinics Program Prevalence Study. Circulation. 1980 Nov:62(4 Pt 2):IV77-82
[PubMed PMID: 6106533]
[17]
Bagatell CJ, Heiman JR, Matsumoto AM, Rivier JE, Bremner WJ. Metabolic and behavioral effects of high-dose, exogenous testosterone in healthy men. The Journal of clinical endocrinology and metabolism. 1994 Aug:79(2):561-7
[PubMed PMID: 8045977]
[18]
Zwald ML, Akinbami LJ, Fakhouri TH, Fryar CD. Prevalence of Low High-density Lipoprotein Cholesterol Among Adults, by Physical Activity: United States, 2011-2014. NCHS data brief. 2017 Mar:(276):1-8
[PubMed PMID: 28282020]
[19]
Toth PP. Reverse cholesterol transport: high-density lipoprotein's magnificent mile. Current atherosclerosis reports. 2003 Sep:5(5):386-93
[PubMed PMID: 12911849]
[20]
Vaisar T, Pennathur S, Green PS, Gharib SA, Hoofnagle AN, Cheung MC, Byun J, Vuletic S, Kassim S, Singh P, Chea H, Knopp RH, Brunzell J, Geary R, Chait A, Zhao XQ, Elkon K, Marcovina S, Ridker P, Oram JF, Heinecke JW. Shotgun proteomics implicates protease inhibition and complement activation in the antiinflammatory properties of HDL. The Journal of clinical investigation. 2007 Mar:117(3):746-56
[PubMed PMID: 17332893]
[21]
Ansell BJ, Fonarow GC, Fogelman AM. High-density lipoprotein: is it always atheroprotective? Current atherosclerosis reports. 2006 Sep:8(5):405-11
[PubMed PMID: 16901411]
[22]
Schaefer EJ, Santos RD, Asztalos BF. Marked HDL deficiency and premature coronary heart disease. Current opinion in lipidology. 2010 Aug:21(4):289-97. doi: 10.1097/MOL.0b013e32833c1ef6. Epub
[PubMed PMID: 20616715]
Level 3 (low-level) evidence
[23]
Schaefer EJ. Clinical, biochemical, and genetic features in familial disorders of high density lipoprotein deficiency. Arteriosclerosis (Dallas, Tex.). 1984 Jul-Aug:4(4):303-22
[PubMed PMID: 6431953]
[24]
Santos RD, Asztalos BF, Martinez LR, Miname MH, Polisecki E, Schaefer EJ. Clinical presentation, laboratory values, and coronary heart disease risk in marked high-density lipoprotein-deficiency states. Journal of clinical lipidology. 2008 Aug:2(4):237-47. doi: 10.1016/j.jacl.2008.06.002. Epub 2008 Jun 13
[PubMed PMID: 21291740]
[25]
Wolinsky H. The effects of beta-adrenergic blocking agents on blood lipid levels. Clinical cardiology. 1987 Oct:10(10):561-6
[PubMed PMID: 2889552]
[26]
Schaefer EJ, Asztalos BF. The effects of statins on high-density lipoproteins. Current atherosclerosis reports. 2006 Jan:8(1):41-9
[PubMed PMID: 16455013]
[27]
Martin G, Duez H, Blanquart C, Berezowski V, Poulain P, Fruchart JC, Najib-Fruchart J, Glineur C, Staels B. Statin-induced inhibition of the Rho-signaling pathway activates PPARalpha and induces HDL apoA-I. The Journal of clinical investigation. 2001 Jun:107(11):1423-32
[PubMed PMID: 11390424]
[28]
Gotto AM Jr, Whitney E, Stein EA, Shapiro DR, Clearfield M, Weis S, Jou JY, Langendörfer A, Beere PA, Watson DJ, Downs JR, de Cani JS. Relation between baseline and on-treatment lipid parameters and first acute major coronary events in the Air Force/Texas Coronary Atherosclerosis Prevention Study (AFCAPS/TexCAPS). Circulation. 2000 Feb 8:101(5):477-84
[PubMed PMID: 10662743]
[29]
Birjmohun RS, Hutten BA, Kastelein JJ, Stroes ES. Efficacy and safety of high-density lipoprotein cholesterol-increasing compounds: a meta-analysis of randomized controlled trials. Journal of the American College of Cardiology. 2005 Jan 18:45(2):185-97
[PubMed PMID: 15653014]
Level 1 (high-level) evidence
[30]
Rubins HB, Robins SJ, Collins D, Fye CL, Anderson JW, Elam MB, Faas FH, Linares E, Schaefer EJ, Schectman G, Wilt TJ, Wittes J. Gemfibrozil for the secondary prevention of coronary heart disease in men with low levels of high-density lipoprotein cholesterol. Veterans Affairs High-Density Lipoprotein Cholesterol Intervention Trial Study Group. The New England journal of medicine. 1999 Aug 5:341(6):410-8
[PubMed PMID: 10438259]
[31]
Tunaru S, Kero J, Schaub A, Wufka C, Blaukat A, Pfeffer K, Offermanns S. PUMA-G and HM74 are receptors for nicotinic acid and mediate its anti-lipolytic effect. Nature medicine. 2003 Mar:9(3):352-5
[PubMed PMID: 12563315]
[32]
Lavie CJ, Mailander L, Milani RV. Marked benefit with sustained-release niacin therapy in patients with "isolated" very low levels of high-density lipoprotein cholesterol and coronary artery disease. The American journal of cardiology. 1992 Apr 15:69(12):1083-5
[PubMed PMID: 1561983]
[33]
Kamanna VS, Kashyap ML. Mechanism of action of niacin. The American journal of cardiology. 2008 Apr 17:101(8A):20B-26B. doi: 10.1016/j.amjcard.2008.02.029. Epub
[PubMed PMID: 18375237]
[34]
AIM-HIGH Investigators, Boden WE, Probstfield JL, Anderson T, Chaitman BR, Desvignes-Nickens P, Koprowicz K, McBride R, Teo K, Weintraub W. Niacin in patients with low HDL cholesterol levels receiving intensive statin therapy. The New England journal of medicine. 2011 Dec 15:365(24):2255-67. doi: 10.1056/NEJMoa1107579. Epub 2011 Nov 15
[PubMed PMID: 22085343]
[35]
Moffatt RJ, Effects of cessation of smoking on serum lipids and high density lipoprotein-cholesterol. Atherosclerosis. 1988 Nov;
[PubMed PMID: 3214483]
[36]
Wood PD, Stefanick ML, Dreon DM, Frey-Hewitt B, Garay SC, Williams PT, Superko HR, Fortmann SP, Albers JJ, Vranizan KM. Changes in plasma lipids and lipoproteins in overweight men during weight loss through dieting as compared with exercise. The New England journal of medicine. 1988 Nov 3:319(18):1173-9
[PubMed PMID: 3173455]
[37]
Wood PD, Stefanick ML, Williams PT, Haskell WL. The effects on plasma lipoproteins of a prudent weight-reducing diet, with or without exercise, in overweight men and women. The New England journal of medicine. 1991 Aug 15:325(7):461-6
[PubMed PMID: 1852180]
[38]
Gordon DJ, Probstfield JL, Garrison RJ, Neaton JD, Castelli WP, Knoke JD, Jacobs DR Jr, Bangdiwala S, Tyroler HA. High-density lipoprotein cholesterol and cardiovascular disease. Four prospective American studies. Circulation. 1989 Jan:79(1):8-15
[PubMed PMID: 2642759]
[39]
Castelli WP, Doyle JT, Gordon T, Hames CG, Hjortland MC, Hulley SB, Kagan A, Zukel WJ. HDL cholesterol and other lipids in coronary heart disease. The cooperative lipoprotein phenotyping study. Circulation. 1977 May:55(5):767-72
[PubMed PMID: 191215]
[40]
Miller NE, Thelle DS, Forde OH, Mjos OD. The Tromsø heart-study. High-density lipoprotein and coronary heart-disease: a prospective case-control study. Lancet (London, England). 1977 May 7:1(8019):965-8
[PubMed PMID: 67464]
Level 2 (mid-level) evidence
[41]
Assmann G, Cullen P, Schulte H. The Münster Heart Study (PROCAM). Results of follow-up at 8 years. European heart journal. 1998 Feb:19 Suppl A():A2-11
[PubMed PMID: 9519336]
[42]
Emerging Risk Factors Collaboration, Di Angelantonio E, Sarwar N, Perry P, Kaptoge S, Ray KK, Thompson A, Wood AM, Lewington S, Sattar N, Packard CJ, Collins R, Thompson SG, Danesh J. Major lipids, apolipoproteins, and risk of vascular disease. JAMA. 2009 Nov 11:302(18):1993-2000. doi: 10.1001/jama.2009.1619. Epub
[PubMed PMID: 19903920]
[43]
Castelli WP. Cholesterol and lipids in the risk of coronary artery disease--the Framingham Heart Study. The Canadian journal of cardiology. 1988 Jul:4 Suppl A():5A-10A
[PubMed PMID: 3179802]
[44]
Castelli WP, Garrison RJ, Wilson PW, Abbott RD, Kalousdian S, Kannel WB. Incidence of coronary heart disease and lipoprotein cholesterol levels. The Framingham Study. JAMA. 1986 Nov 28:256(20):2835-8
[PubMed PMID: 3773200]
[45]
Dong W, Wong KHY, Liu Y, Levy-Sakin M, Hung WC, Li M, Li B, Jin SC, Choi J, Lopez-Giraldez F, Vaka D, Poon A, Chu C, Lao R, Balamir M, Movsesyan I, Malloy MJ, Zhao H, Kwok PY, Kane JP, Lifton RP, Pullinger CR. Whole-exome sequencing reveals damaging gene variants associated with hypoalphalipoproteinemia. Journal of lipid research. 2022 Jun:63(6):100209. doi: 10.1016/j.jlr.2022.100209. Epub 2022 Apr 20
[PubMed PMID: 35460704]
[46]
Viaud M, Abdel-Wahab O, Gall J, Ivanov S, Guinamard R, Sore S, Merlin J, Ayrault M, Guilbaud E, Jacquel A, Auberger P, Wang N, Levine RL, Tall AR, Yvan-Charvet L. ABCA1 Exerts Tumor-Suppressor Function in Myeloproliferative Neoplasms. Cell reports. 2020 Mar 10:30(10):3397-3410.e5. doi: 10.1016/j.celrep.2020.02.056. Epub
[PubMed PMID: 32160545]
[47]
Hollmén M, Figueiredo CR, Jalkanen S. New tools to prevent cancer growth and spread: a 'Clever' approach. British journal of cancer. 2020 Aug:123(4):501-509. doi: 10.1038/s41416-020-0953-0. Epub 2020 Jun 29
[PubMed PMID: 32595212]
[48]
Cancer Genome Atlas Research Network. Electronic address: wheeler@bcm.edu, Cancer Genome Atlas Research Network. Comprehensive and Integrative Genomic Characterization of Hepatocellular Carcinoma. Cell. 2017 Jun 15:169(7):1327-1341.e23. doi: 10.1016/j.cell.2017.05.046. Epub
[PubMed PMID: 28622513]
[49]
Ladanyi A, Mukherjee A, Kenny HA, Johnson A, Mitra AK, Sundaresan S, Nieman KM, Pascual G, Benitah SA, Montag A, Yamada SD, Abumrad NA, Lengyel E. Adipocyte-induced CD36 expression drives ovarian cancer progression and metastasis. Oncogene. 2018 Apr:37(17):2285-2301. doi: 10.1038/s41388-017-0093-z. Epub 2018 Feb 5
[PubMed PMID: 29398710]
[50]
Langlois B, Perrot G, Schneider C, Henriet P, Emonard H, Martiny L, Dedieu S. LRP-1 promotes cancer cell invasion by supporting ERK and inhibiting JNK signaling pathways. PloS one. 2010 Jul 14:5(7):e11584. doi: 10.1371/journal.pone.0011584. Epub 2010 Jul 14
[PubMed PMID: 20644732]
[51]
Cui Y, Liang S, Zhang S, Zhang C, Zhao Y, Wu D, Wang J, Song R, Wang J, Yin D, Liu Y, Pan S, Liu X, Wang Y, Han J, Meng F, Zhang B, Guo H, Lu Z, Liu L. ABCA8 is regulated by miR-374b-5p and inhibits proliferation and metastasis of hepatocellular carcinoma through the ERK/ZEB1 pathway. Journal of experimental & clinical cancer research : CR. 2020 May 19:39(1):90. doi: 10.1186/s13046-020-01591-1. Epub 2020 May 19
[PubMed PMID: 32430024]
[52]
Wong M, Funasaka K, Obayashi T, Miyahara R, Hirooka Y, Hamaguchi M, Goto H, Senga T. AMPD3 is associated with the malignant characteristics of gastrointestinal stromal tumors. Oncology letters. 2017 Mar:13(3):1281-1287. doi: 10.3892/ol.2016.5532. Epub 2016 Dec 23
[PubMed PMID: 28454247]
Level 3 (low-level) evidence
[53]
Fernandez P, Carretero J, Medina PP, Jimenez AI, Rodriguez-Perales S, Paz MF, Cigudosa JC, Esteller M, Lombardia L, Morente M, Sanchez-Verde L, Sotelo T, Sanchez-Cespedes M. Distinctive gene expression of human lung adenocarcinomas carrying LKB1 mutations. Oncogene. 2004 Jun 24:23(29):5084-91
[PubMed PMID: 15077168]
[54]
Reiss K, Del Valle L, Lassak A, Trojanek J. Nuclear IRS-1 and cancer. Journal of cellular physiology. 2012 Aug:227(8):2992-3000. doi: 10.1002/jcp.24019. Epub
[PubMed PMID: 22454254]
[55]
Pedersen KM, Çolak Y, Bojesen SE, Nordestgaard BG. Low high-density lipoprotein and increased risk of several cancers: 2 population-based cohort studies including 116,728 individuals. Journal of hematology & oncology. 2020 Sep 30:13(1):129. doi: 10.1186/s13045-020-00963-6. Epub 2020 Sep 30
[PubMed PMID: 32998735]
[56]
Downs JR, Clearfield M, Weis S, Whitney E, Shapiro DR, Beere PA, Langendorfer A, Stein EA, Kruyer W, Gotto AM Jr. Primary prevention of acute coronary events with lovastatin in men and women with average cholesterol levels: results of AFCAPS/TexCAPS. Air Force/Texas Coronary Atherosclerosis Prevention Study. JAMA. 1998 May 27:279(20):1615-22
[PubMed PMID: 9613910]