Introduction
Acute generalized exanthematous pustulosis (AGEP) is a rare cutaneous adverse reaction in which tiny nonfollicular sterile pustules with underlying erythema develop and spread rapidly.[1] Initially, AGEP was thought to be a variant of pustular psoriasis until it was described and named as a distinct clinical entity in 1980.[2]
Most cases of AGEP occur as an adverse drug reaction, but cases have also been reported following infectious insults or contact with physical triggers. Although AGEP is often self-limited, it may lead to systemic complications; identification and discontinuation of the triggering agent is the mainstay of treatment.[3][4]
Generally classified alongside other severe cutaneous adverse reactions (SCARs) as a type IV hypersensitivity reaction, confirmation of the full breadth of the pathophysiology of AGEP remains under investigation.[5]
Etiology
The vast majority of reported cases of AGEP are due to an adverse drug reaction, with several cases confirmed via patch testing. Systemic antibiotics are the most commonly implicated agents, particularly beta-lactam antibiotics and macrolides. Other drugs reported to trigger AGEP include hydroxychloroquine, antifungal, antiviral, and antiparasitic drugs, as well as antineoplastic and antirheumatic agents, analgesics, anticonvulsants, and intravenous contrast media, among others. In some cases, even corticosteroids have been reported as the offending drugs.[3][6]
AGEP has been associated with various triggers, including infections, spider bites, contact allergens, herbal remedies, and psoralen-UVA treatment. However, the cause of some cases remains unknown or lacks sufficient confirmation to identify a specific trigger.[4][6]
Certain medications are reported as the suspected cause of AGEP.[7][8][9] These include, but are not limited to, the following:
| Antibiotics |
Beta-lactams, macrolides, quinolones, tetracyclines, antituberculosis agents, sulfonamides |
| Other anti-infectives |
Antifungals, antimalarials, antiprotozoals, antivirals, antihelmintic agents |
| Analgesics |
NSAIDs, opioids, acetaminophen, muscle relaxants |
| Nervous system medications |
Anticonvulsants, antidepressants, mood stabilizers, antipsychotics |
| Cardiovascular medications |
Calcium channel blockers, beta-blockers, ACE inhibitors, RAAS modulators, diuretics, lipid-lowering agents, antiplatelets |
| Respiratory medications |
Leukotriene receptor antagonists |
| Antineoplastic |
Kinase inhibitors, monoclonal antibodies, immunomodulators, chemotherapeutic agents |
| Other |
Metformin, proton-pump inhibitors, antihistamines, contrast media, steroids, topical agents, mercury, herbal remedies, vitamins, vaccines, anti-anemic preparations, anesthetics |
Epidemiology
AGEP is considered a rare condition, with an estimated incidence ranging from one to five cases per million per year.[6] However, cases of AGEP could be underreported due to the clinical overlap with other cutaneous drug eruptions and the self-limited nature of most cases that may resolve before referral to a dermatologist.[10][11]
The age range of patients with AGEP is broad, as seen in a 2022 case series of 297 patients that included patients from 1 to 93 years old, with patients over 25 most heavily represented, specifically those between 40 and 64 years of age. Similar to other drug-induced SCARs, AGEP has a female predominance. Women account for approximately 65-80% of reported cases.[9]
The onset and resolution of AGEP vary between causative medications with different pharmacokinetics; as such, the female predominance of AGEP may be partly due to differences in drug metabolism compared to men.[12][13] Given that over 85-90% of cases occur in the setting of drug exposure, the geographic distribution seems to align with countries with higher availability and clinical use of medications, such as pristinamycin in France, known to cause AGEP.[14][15]
Other reported correlations with cases of AGEP include patients with HLA-B51, HLA-DR-11, and HLA-DQ3 genotypes associated with T-cell activation, as well as IL-36 receptor antagonist gene mutations resulting in increased IL-36 signaling.[5]
Pathophysiology
The pathogenesis of AGEP is partially, but not yet fully, understood. AGEP is typically described as a T-cell-mediated type IV hypersensitivity reaction with neutrophilic inflammation. After exposure to a triggering factor, such as a new medication, host proteins bind drug metabolites to form an epitope captured by antigen-presenting cells. Drug-specific CD4+ and CD8+ cells are then activated and migrate to the skin, then cytotoxic proteins (perforin, granzyme B, Fas ligand) are released to induce keratinocyte apoptosis. This creates vesicles that become sterile pustules as neutrophils are recruited via CXCL8 and IL-8. Interferon-gamma (IFN gamma) propagates additional neutrophil recruitment via additional CXCL8, while granulocyte-macrophage colony-stimulating factor (GM-CSF) protects the neutrophils from apoptosis.[5]
The innate immune system's involvement has also been implicated based on elevated IL-17 and IL-22 in peripheral blood samples of AGEP patients. Th17 cells produce IL-17 and IL-22, which stimulate additional IL-8 production from keratinocytes, thereby propagating neutrophil recruitment and pustule formation.[7][16]
Furthermore, IL-36 gene mutations resulting in dysregulated IL-36 signaling have been associated with AGEP, implying participation of the IL-36 pro-inflammatory cytokine cascade in the propagation of this condition.[10] Increased IL-4 and IL-5 cytokine production have also been reported in some cases, which could explain the peripheral eosinophilia and eosinophilic infiltrates in AGEP skin.[5] Overall, additional studies are needed to elucidate the pathogenesis of AGEP further.
Histopathology
Histopathologic findings consistent with AGEP include non-follicular spongiform pustules within the epidermis, particularly within and below the stratum corneum (causing a subcorneal dermatosis), edema of the papillary dermis with neutrophilic and eosinophilic perivascular infiltrates.[5][8] Additional features include necrotic keratinocytes, the potential confluence between the intraepidermal and subcorneal pustules, and mixed inflammatory dermal and interstitial infiltrates.[6][10]
Specifically, when differentiating between pustular psoriasis and AGEP, AGEP has necrotic keratinocytes, spongiosis of the epidermis, mixed neutrophilic and eosinophilic infiltrates, and lacks the dilated blood vessels within the dermal papillae that are classically described in psoriasis.[11][12]
Although not typically performed in the analysis of skin biopsies, gene expression profiling of AGEP skin shows increased IL36G expression compared to healthy skin, and immunohistochemical analysis shows a strong presence of IL-36 alpha and IL-36 gamma in the pustules as well as the peri-pustular epidermis of AGEP biopsies.[7]
History and Physical
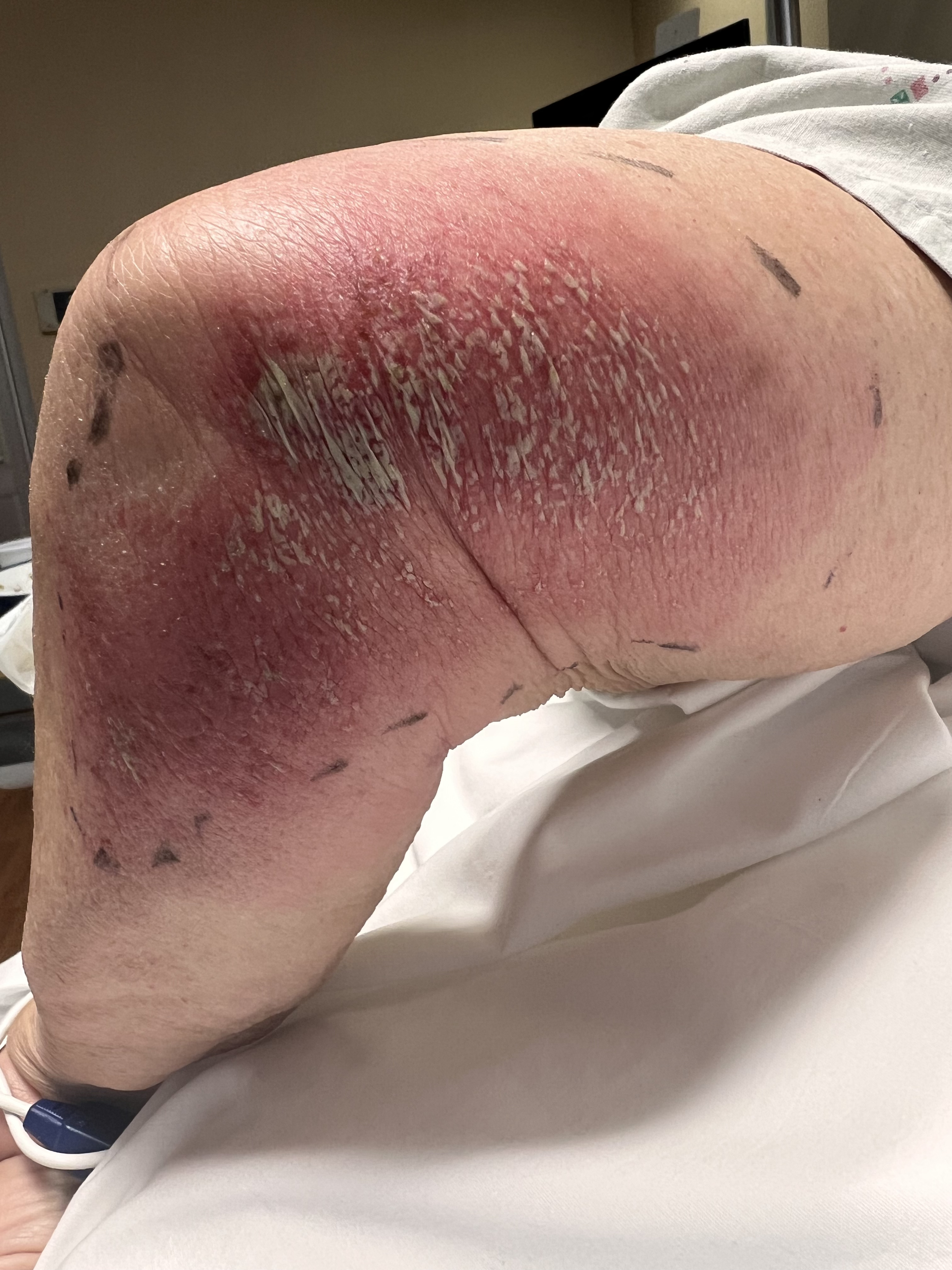
The appearance of AGEP is described as tiny nonfollicular sterile pustules with underlying erythema that develop and spread rapidly and are accompanied by pruritus or burning sensation plus an actual or subjective fever. Although often described as “pinhead” size, the pustules may coalesce, appear more prominent, and lead to superficial skin detachment with gentle pressure akin to a positive Nikolsky sign.[17]
Symptoms begin in flexural or intertriginous regions before becoming more generalized to the trunk, limbs, and face within a few hours to days while usually sparing palms and soles.[6][10] Mucosal surfaces are not typically involved, but when they are, this involvement is often restricted to the oral and buccal mucosa.[17]
Over the next 1 to 2 weeks, the pustules desquamate in a collaret-like manner. Other reported symptoms include lymphadenopathy or atypical presentations that include purpura, vesicles, bullae, or symptoms localized to the face, neck, and chest. Historical clues consistent with AGEP include rapid development of symptoms, often within ten days or less of exposure to an offending medication, and resolution within 15 days of discontinuation.[3][8]
In some cases, such as AGEP triggered by hydroxychloroquine or terbinafine, the time from onset to resolution may be much longer.[12][18]
Evaluation
In addition to a detailed history, physical exam, and medication reconciliation, a punch biopsy that includes a pustule and blood tests is useful to aid in diagnosis and to evaluate for systemic involvement.[11] Useful lab tests include a complete blood count (CBC), a comprehensive metabolic panel (CMP) to include renal and liver function tests (LFT), and C-reactive protein (CRP).[17]
The presence or absence of other symptoms and hemodynamic stability of a patient will dictate the urgency and setting (outpatient versus inpatient) in which this workup occurs. Suppose a patient's clinical presentation overlaps with other severe cutaneous adverse reactions, such as Stevens-Johnson syndrome or toxic epidermal necrolysis. In that case, they may require an evaluation and care at a specialized burn unit.[19]
A scoring system published in 2001 by Sidoroff et al., often called the EuroSCAR criteria or AGEP validation score, includes clinical information, lab results, and histopathologic findings. These criteria stratify cases into the likelihood that AGEP is the underlying diagnosis.
According to these metrics, a score of 0 or below rules out the possibility of AGEP. A score of 1 to 4 cannot rule out AGEP, whereas a score of 5 to 7 indicates that AGEP is probable. A score of 8 to 12 is considered to diagnose AGEP definitively. These criteria are shown.[6][8]
Exanthem Features
- No pustules or unable to determine (0), Pustules present and equivocal (+1), Pustules present in significant number, nonfollicular, and minuscule/"pinhead" < 5 mm size, can be confluent (+2).
- No erythema or unable to determine (0), Erythema present and equivocal (+1), Erythema present and widespread as a base upon which the pustules are located (+2).
- Unable to determine distribution pattern (0), Distribution pattern equivocal (+1), Distribution begins in intertriginous regions or face with rapid spread to trunk and limbs (+2).
- No post-pustular desquamation or unable to determine (0), Post-pustular desquamation (+1).
Other Features
- Mucosal involvement (- 2), no mucosal involvement (0).
- Onset > 10 days (-2), onset <= 10 days (0).
- Resolution > 15 days (-4), resolution <= 15 days (0)
- Temperature < 38 degrees C (100.4 degrees F) (0), Fever >- 38 degrees C (100.4 degrees F) (+1).
- WBC < 7,000 cells/mm^3 (0), WBC >= 7,000 cells/mm^3 (+1)
Histologic Features
- Consistent with another diagnosis (-10).
- Not representative or no histology (0).
- Peripheral neutrophil exocytosis (+1).
- Non-spongiform subcorneal and/or intraepidermal or nonspecified pustules with papillary edema (+2), Spongiform subcorneal and/or intraepidermal or nonspecific pustules without papillary edema (+2).
- Spongiform subcorneal and/or intraepidermal pustules with papillary edema (+3).
In addition to clinical evaluation and biopsy, attempting to confirm the culprit medication may be appropriate in cases of polypharmacy. Any diagnostic tests that involve symptom provocation should not be attempted until at least six weeks after the complete resolution of symptoms. Patch testing is the preferred approach, with intradermal or prick tests potentially useful if patch testing is negative and the benefits outweigh the risks.[6] The estimated sensitivity of patch testing for AGEP is up to 58%; therefore, a negative patch test result cannot rule out a potential triggering agent.[20]
Ex vivo tests such as the lymphocyte transformation test (LTT) and enzyme-linked immunospot assay (ELISpot) have potential utility but currently are limited in their clinical use. One potential benefit is that these tests do not rely on re-creating the patient's symptoms and may be more desirable for patient comfort. Drawbacks of these tests include lack of availability at many healthcare facilities, the requirement for well-preserved and viable T-lymphocytes from the patient, and wide variation in sensitivity for delayed hypersensitivity reactions. The reported sensitivity is between 27% and 74% for LLT and 35% to 85% for ELISpot.[21]
Treatment / Management
Treatment involves discontinuing the offending agent and providing supportive care such as topical ceramides and prevention of secondary infection with proper skin hygiene. Additional supportive treatments include topical corticosteroids, antipyretics, and antihistamines.[5][21]
Treatments reported for some inpatient cases include systemic corticosteroids, cyclosporine, acitretin, dapsone, infliximab, and intravenous immune globulin (IVIG).[4] In cases of severe or refractory AGEP, there may be a role for systemic steroids or cyclosporine, with the potential to escalate to infliximab and intravenous immune globulin; however, these treatments are not routinely indicated. Evidence-based guidelines for AGEP that fail to respond to supportive therapy do not yet exist, as current recommendations are based on case studies without controlled studies.[5]
For patients with a history of AGEP, empiric avoidance of the suspected trigger and appropriate documentation of an adverse drug reaction history in the patient’s medical record is appropriate.[6]
Differential Diagnosis
Since the course of AGEP has pustular and desquamative phases, the differential diagnosis includes pustular psoriasis, subcorneal pustular dermatosis (Sneddon-Wilkinson disease), varicelliform eruption, pustular vasculitis, staphylococcal scalded skin syndrome, pustular erythema multiforme, bullous impetigo, bullous tinea, pemphigus foliaceous, pemphigus erythematosus, pemphigus vulgaris, and other severe cutaneous adverse reactions and drug eruptions such as Stevens-Johnson syndrome (SJS), toxic epidermal necrolysis (TEN), or drug reaction with eosinophilia and systemic symptoms (DRESS).[5][8][17] Pustular psoriasis is typically the most difficult to differentiate from AGEP.
Prognosis
The prognosis for AGEP is excellent, and symptoms most often improve or completely resolve within 15 days of medication discontinuation. Still, the recovery timeline may vary depending on the specific pharmacokinetics of the causative agent. The long half-life of hydroxychloroquine is thought to explain prolonged symptoms of AGEP induced by this drug compared to other cases.[12]
Identifying a suitable alternative treatment is crucial for a patient's overall well-being if AGEP is caused by a medication they would otherwise take long-term, such as first-line medications for cardiovascular disease, inflammatory arthritis, or epilepsy.
The previously cited case-fatality rate of 2 to 5% cannot be definitively attributed solely to AGEP without other comorbidities and is therefore disputed.[4] Recent literature reviews have reported fatalities among patients with AGEP. However, these fatalities were observed in patients whose health was tenuous before the onset of AGEP. Additionally, some patients' AGEP had resolved before their death. Most fatalities were ultimately attributed to sepsis or multi-organ failure.[3]
Complications
Reported complications of AGEP include bacterial superinfection of desquamating skin, hepatomegaly, lymphadenopathy, liver injury, kidney injury, hypocalcemia, pleural effusions, respiratory distress, agranulocytosis, and even multiorgan involvement. However, reporting bias and retrospective hospital patient data likely influence the reported frequencies.[17][22]
One review of 340 AGEP cases in the United States found roughly 8% of patients had acute kidney insult as defined by serum creatinine 1.5 times their baseline, and 8.5% had elevated liver enzymes at least twice the upper limit of normal. In severe or atypical cases of AGEP refractory to supportive care and medication discontinuation alone, AGEP may mimic septic shock or clinically overlap with TEN or DRESS, with multiorgan involvement requiring additional treatment interventions. Complications are rare, and most cases resolve after stopping the offending medication.[10]
Deterrence and Patient Education
Patients can be reassured that AGEP often resolves by discontinuing the causative agent, time, and symptom-guided therapy. The previously cited case-fatality rate of 2 to 5% cannot be definitively attributed solely to AGEP without other comorbidities.[3][4] Topical steroids, secondary skin infection prevention, and topical moisturizers to rehydrate skin during desquamation are often sufficient treatment measures.[11]
If the cause of AGEP is ambiguous, patch or intradermal testing may be considered at least six weeks after complete resolution. However, empiric avoidance of the suspected trigger and appropriate documentation of an adverse drug reaction history in the patient's medical record, without confirmatory testing, is also reasonable.[6]
Enhancing Healthcare Team Outcomes
Although AGEP is predominantly self-limited and resolves with discontinuation and avoidance of the triggering agent, the rapid progression of symptoms with associated physical discomfort and potential for secondary infection without proper skincare can be particularly distressing for patients. Lack of proper diagnosis may delay discontinuation of the causative agent or potentially lead to treatment with systemic corticosteroids despite limited evidence that they expedite recovery or shorten disease duration.[6]
On the other hand, patients with mild or localized AGEP due to a medication that is otherwise critical to their health with no suitable alternative may be appropriate for continuing the offending drug. For patients to make an informed decision in this hypothetical scenario, further data is needed to clarify the pathogenesis, appropriately characterize the risks of repeated exposures, and develop evidence-based treatment guidelines.
Collaboration between an interprofessional team of dermatologists, internists, critical care physicians, immunologists, nurses, and other healthcare specialists in clinical and academic settings is required to enhance patient-centered care. Clinical recognition and publication of cases of AGEP in both the inpatient and outpatient setting, controlled trials of specific treatments in severe or refractory cases, and detailed immunologic studies are needed to understand this condition better, clarify the details of its pathogenesis, and establish evidence-based treatment guidelines. Expanding the knowledge base of AGEP will increase diagnostic fidelity, improve patient counseling and reassurance, and develop effective treatment options.