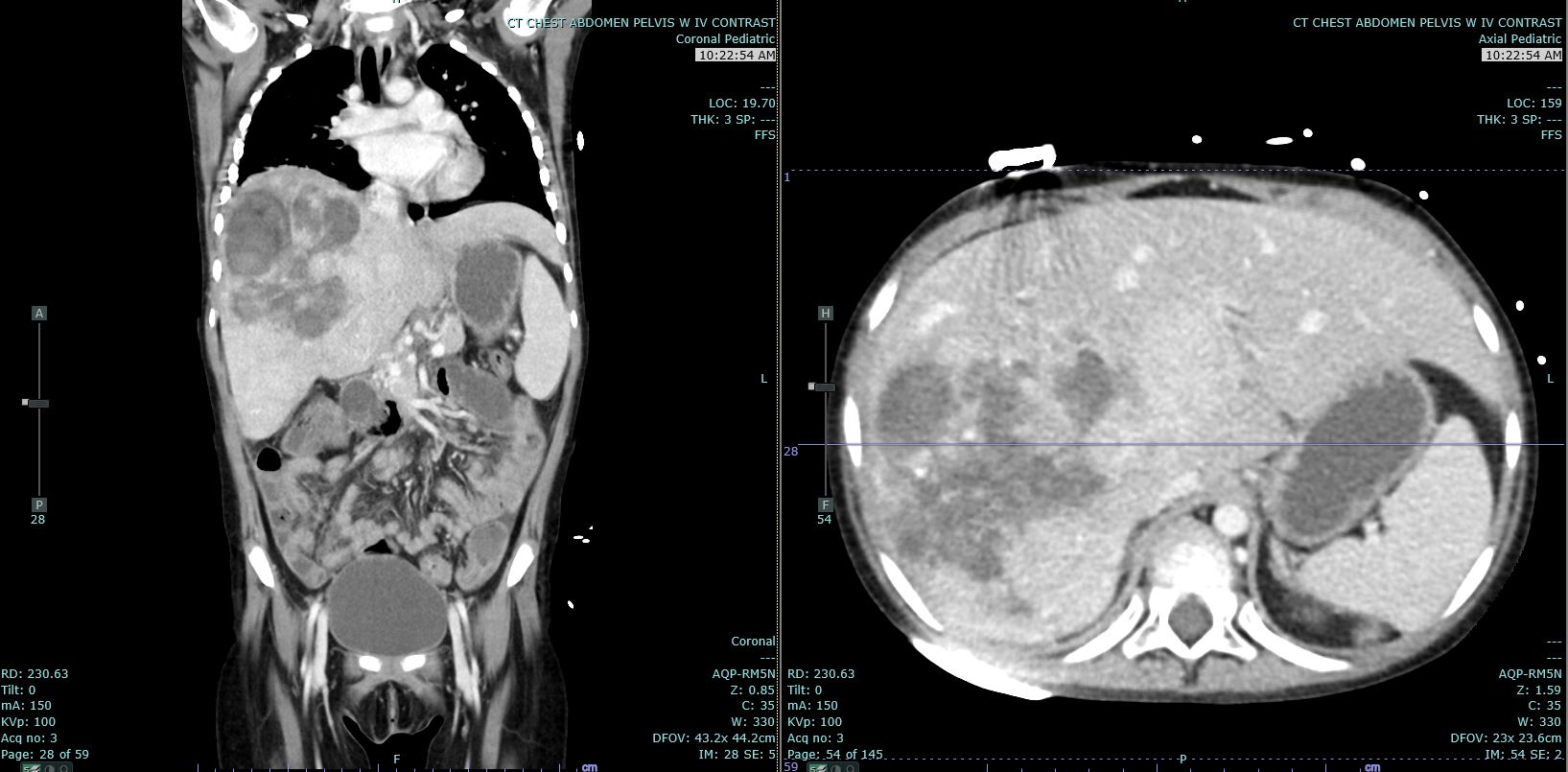
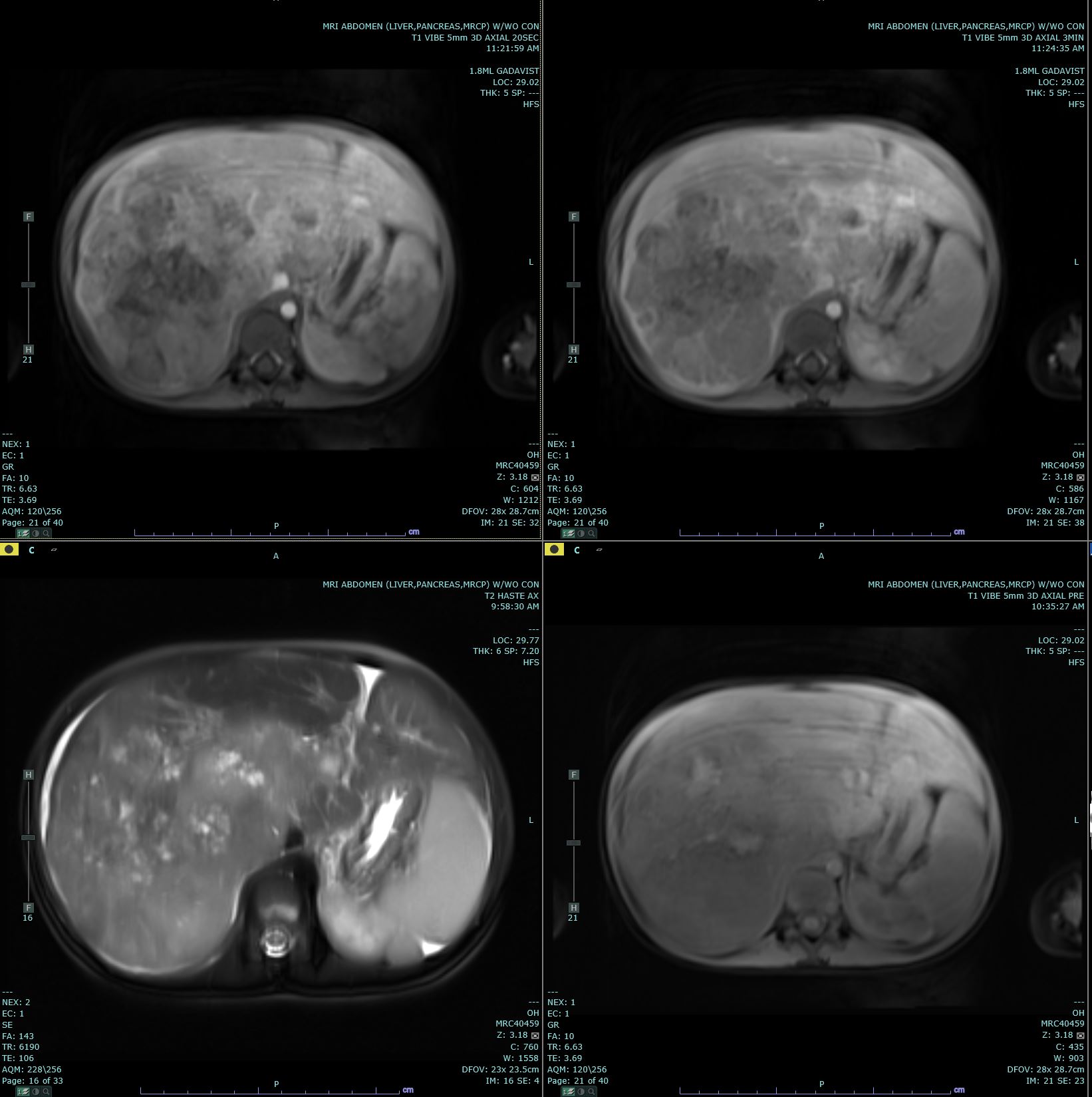
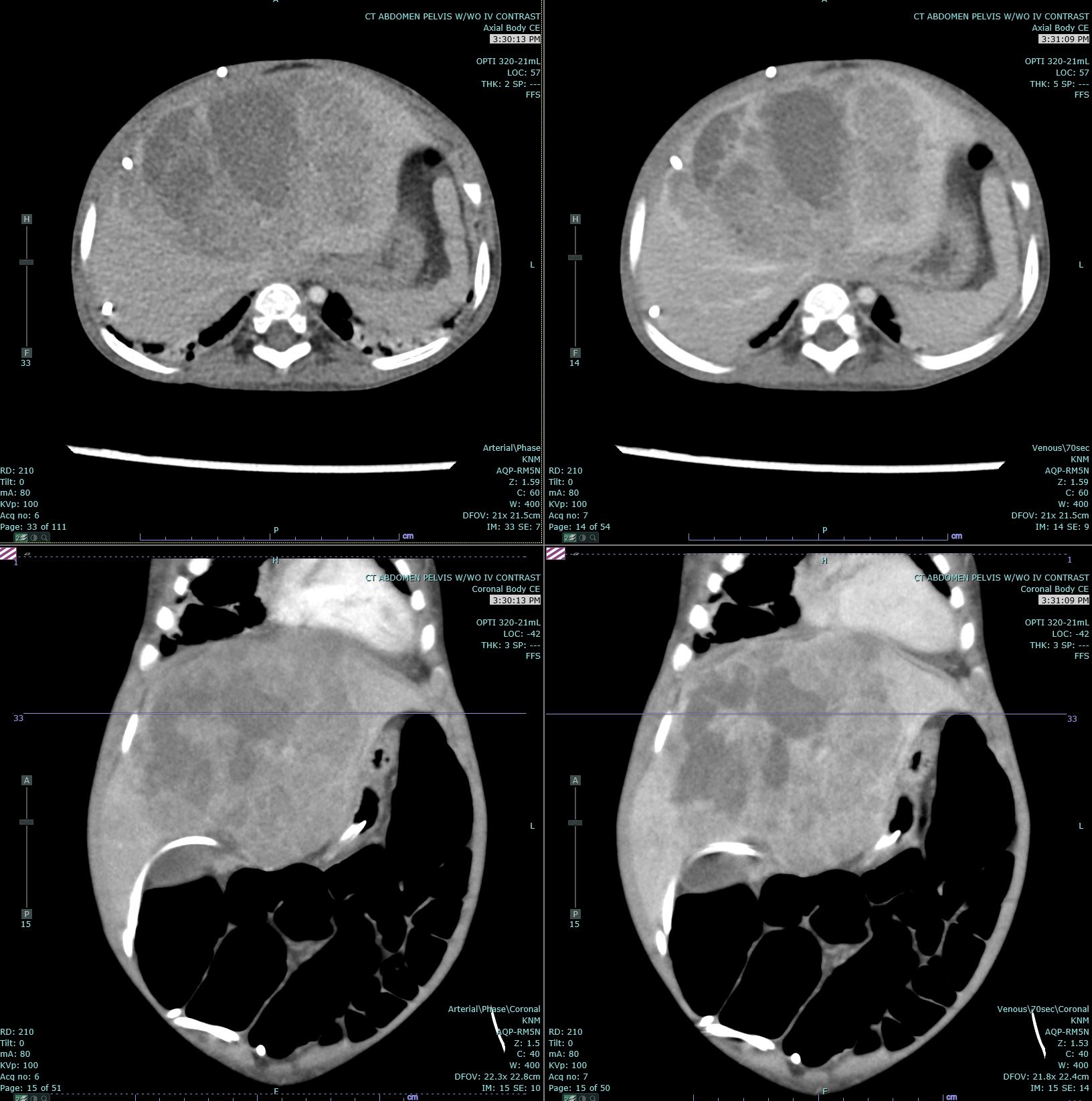
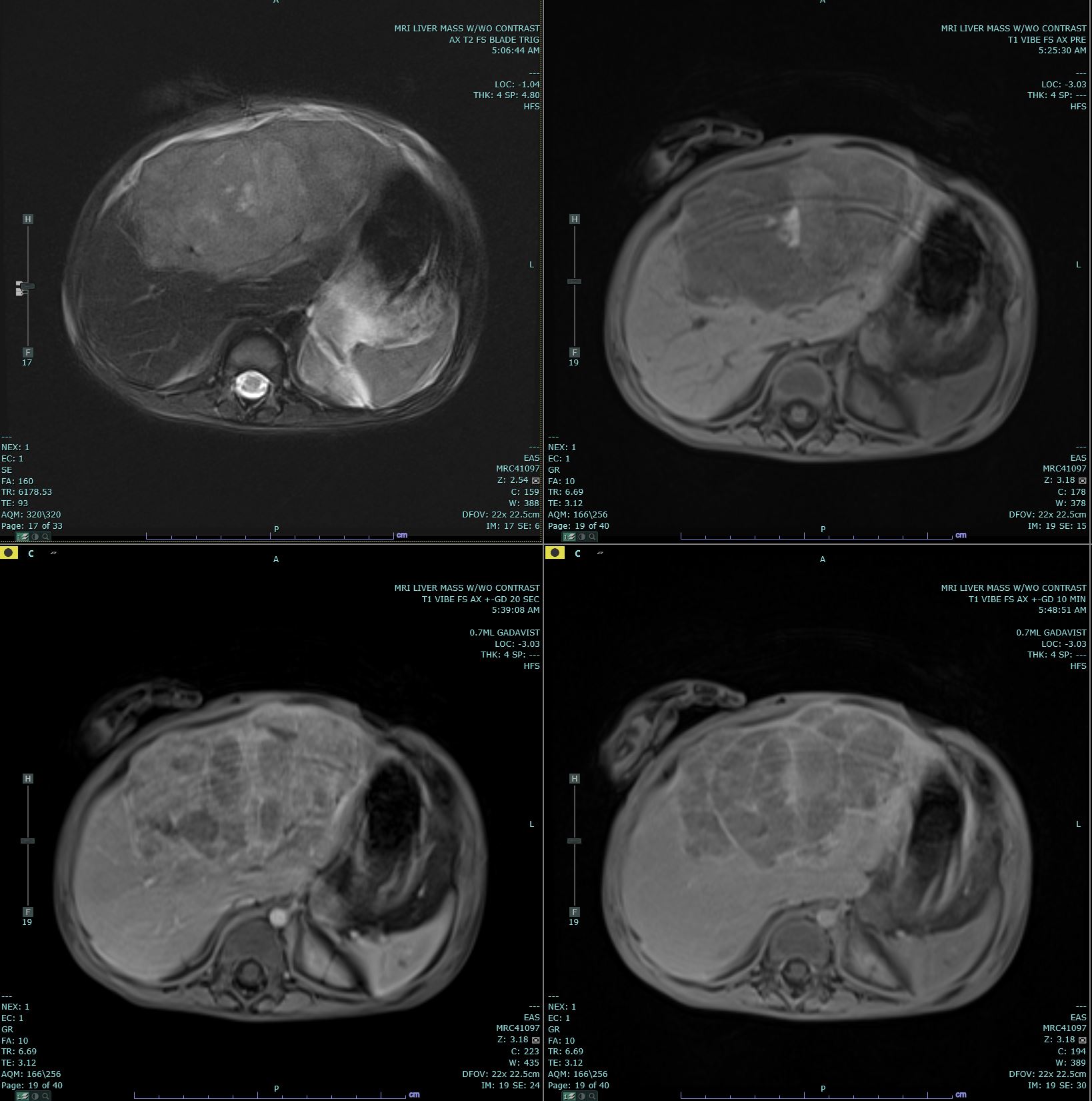
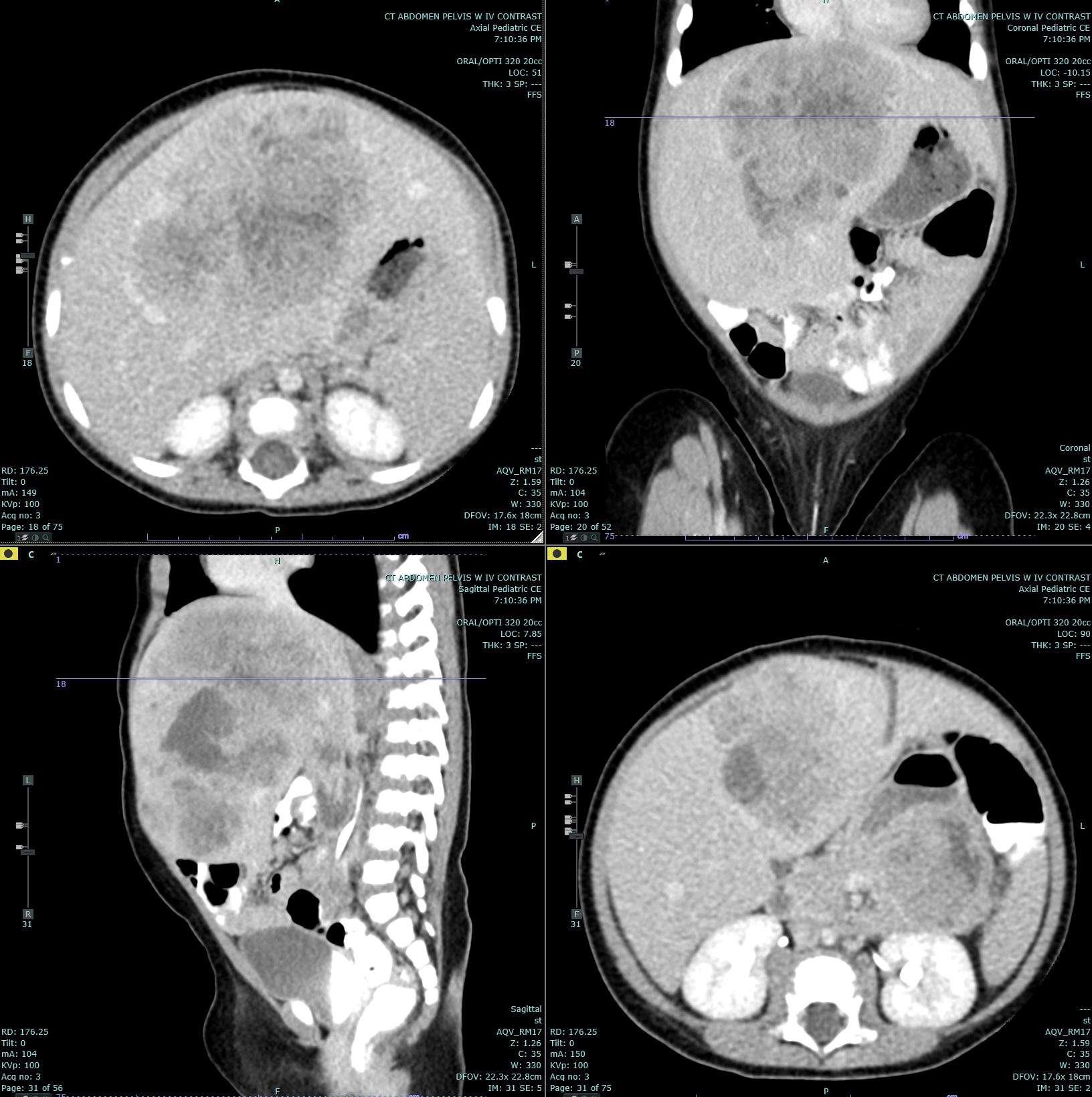
[1]
Sharma D, Subbarao G, Saxena R. Hepatoblastoma. Seminars in diagnostic pathology. 2017 Mar:34(2):192-200. doi: 10.1053/j.semdp.2016.12.015. Epub 2016 Dec 23
[PubMed PMID: 28126357]
[2]
Czauderna P, Lopez-Terrada D, Hiyama E, Häberle B, Malogolowkin MH, Meyers RL. Hepatoblastoma state of the art: pathology, genetics, risk stratification, and chemotherapy. Current opinion in pediatrics. 2014 Feb:26(1):19-28. doi: 10.1097/MOP.0000000000000046. Epub
[PubMed PMID: 24322718]
Level 3 (low-level) evidence
[3]
Finegold MJ, Lopez-Terrada DH, Bowen J, Washington MK, Qualman SJ, College of American Pathologists. Protocol for the examination of specimens from pediatric patients with hepatoblastoma. Archives of pathology & laboratory medicine. 2007 Apr:131(4):520-9
[PubMed PMID: 17425379]
[4]
Heck JE, Meyers TJ, Lombardi C, Park AS, Cockburn M, Reynolds P, Ritz B. Case-control study of birth characteristics and the risk of hepatoblastoma. Cancer epidemiology. 2013 Aug:37(4):390-5. doi: 10.1016/j.canep.2013.03.004. Epub 2013 Apr 1
[PubMed PMID: 23558166]
Level 2 (mid-level) evidence
[5]
Curia MC, Zuckermann M, De Lellis L, Catalano T, Lattanzio R, Aceto G, Veschi S, Cama A, Otte JB, Piantelli M, Mariani-Costantini R, Cetta F, Battista P. Sporadic childhood hepatoblastomas show activation of beta-catenin, mismatch repair defects and p53 mutations. Modern pathology : an official journal of the United States and Canadian Academy of Pathology, Inc. 2008 Jan:21(1):7-14
[PubMed PMID: 17962810]
[6]
Ng K, Rana A, Masand P, Patel K, Heczey A, Goss J, Himes R. Fatal Central Nervous System Post-Transplant Lymphoproliferative Disease in a Patient Who Underwent Liver Transplantation for Hepatoblastoma. Journal of pediatric gastroenterology and nutrition. 2018 Jan:66(1):e21-e23. doi: 10.1097/MPG.0000000000001725. Epub
[PubMed PMID: 28837514]
[7]
Zhong S, Zhao Y, Fan C. Hepatoblastoma with pure fetal epithelial differentiation in a 10-year-old boy: A rare case report and review of the literature. Medicine. 2018 Jan:97(2):e9647. doi: 10.1097/MD.0000000000009647. Epub
[PubMed PMID: 29480877]
Level 3 (low-level) evidence
[8]
Pateva IB, Egler RA, Stearns DS. Hepatoblastoma in an 11-year-old: Case report and a review of the literature. Medicine. 2017 Jan:96(2):e5858. doi: 10.1097/MD.0000000000005858. Epub
[PubMed PMID: 28079820]
Level 3 (low-level) evidence
[9]
López-Terrada D, Alaggio R, de Dávila MT, Czauderna P, Hiyama E, Katzenstein H, Leuschner I, Malogolowkin M, Meyers R, Ranganathan S, Tanaka Y, Tomlinson G, Fabrè M, Zimmermann A, Finegold MJ, Children's Oncology Group Liver Tumor Committee. Towards an international pediatric liver tumor consensus classification: proceedings of the Los Angeles COG liver tumors symposium. Modern pathology : an official journal of the United States and Canadian Academy of Pathology, Inc. 2014 Mar:27(3):472-91. doi: 10.1038/modpathol.2013.80. Epub 2013 Sep 6
[PubMed PMID: 24008558]
Level 3 (low-level) evidence
[10]
Hiyama E. Pediatric hepatoblastoma: diagnosis and treatment. Translational pediatrics. 2014 Oct:3(4):293-9. doi: 10.3978/j.issn.2224-4336.2014.09.01. Epub
[PubMed PMID: 26835349]
[11]
Meyers RL, Tiao G, de Ville de Goyet J, Superina R, Aronson DC. Hepatoblastoma state of the art: pre-treatment extent of disease, surgical resection guidelines and the role of liver transplantation. Current opinion in pediatrics. 2014 Feb:26(1):29-36. doi: 10.1097/MOP.0000000000000042. Epub
[PubMed PMID: 24362406]
Level 3 (low-level) evidence
[12]
Uchida H, Sakamoto S, Sasaki K, Takeda M, Hirata Y, Fukuda A, Hishiki T, Irie R, Nakazawa A, Miyazaki O, Nosaka S, Kasahara M. Surgical treatment strategy for advanced hepatoblastoma: Resection versus transplantation. Pediatric blood & cancer. 2018 Dec:65(12):e27383. doi: 10.1002/pbc.27383. Epub 2018 Aug 7
[PubMed PMID: 30084209]
[13]
Meyers RL, Maibach R, Hiyama E, Häberle B, Krailo M, Rangaswami A, Aronson DC, Malogolowkin MH, Perilongo G, von Schweinitz D, Ansari M, Lopez-Terrada D, Tanaka Y, Alaggio R, Leuschner I, Hishiki T, Schmid I, Watanabe K, Yoshimura K, Feng Y, Rinaldi E, Saraceno D, Derosa M, Czauderna P. Risk-stratified staging in paediatric hepatoblastoma: a unified analysis from the Children's Hepatic tumors International Collaboration. The Lancet. Oncology. 2017 Jan:18(1):122-131. doi: 10.1016/S1470-2045(16)30598-8. Epub 2016 Nov 22
[PubMed PMID: 27884679]
[14]
Dall'Igna P, Brugieres L, Christin AS, Maibach R, Casanova M, Alaggio R, de Goyet JV, Zsiros J, Morland B, Czauderna P, Childs M, Aronson DC, Branchereau S, Brock P, Perilongo G. Hepatoblastoma in children aged less than six months at diagnosis: A report from the SIOPEL group. Pediatric blood & cancer. 2018 Jan:65(1):. doi: 10.1002/pbc.26791. Epub 2017 Sep 18
[PubMed PMID: 28921839]
[15]
Kiruthiga KG, Ramakrishna B, Saha S, Sen S. Histological and immunohistochemical study of hepatoblastoma: correlation with tumour behaviour and survival. Journal of gastrointestinal oncology. 2018 Apr:9(2):326-337. doi: 10.21037/jgo.2018.01.08. Epub
[PubMed PMID: 29755772]
[16]
Wu JF, Chang HH, Lu MY, Jou ST, Chang KC, Ni YH, Chang MH. Prognostic roles of pathology markers immunoexpression and clinical parameters in Hepatoblastoma. Journal of biomedical science. 2017 Aug 29:24(1):62. doi: 10.1186/s12929-017-0369-1. Epub 2017 Aug 29
[PubMed PMID: 28851352]
[17]
Shanmugam N, Scott JX, Kumar V, Vij M, Ramachandran P, Narasimhan G, Reddy MS, Kota V, Munirathnam D, Kelgeri C, Sundaram K, Rela M. Multidisciplinary management of hepatoblastoma in children: Experience from a developing country. Pediatric blood & cancer. 2017 Mar:64(3):. doi: 10.1002/pbc.26249. Epub 2016 Oct 26
[PubMed PMID: 27781375]