Continuing Education Activity
Laugier-Hunziker syndrome, also known as Laugier-Gerbig-Hunziker syndrome or Laugier-Hunziker-Baran syndrome or idiopathic lenticular mucocutaneous pigmentation, is a hereditary pigmentary disorder characterized by a unique expression of pigmentation over the mucosal, nail, and acral sites. It is commonly mistaken for Addison's disease, Peutz Jeghers syndrome, Cronkhite-Canada syndrome. The condition is known to be benign nevertheless a few associations with esophageal melanocytosis, actinic lichen planus, hypocellular bone marrow, and thrombocytopenia have been reported. This activity presents the clinical presentation, evaluation, and management of Laugier-Hunziker syndrome and highlights the role of the interprofessional team in the management of affected patients.
Objectives:
- Describe the etiology of Laugier-Hunziker syndrome.
- Explain the key differentiating features specific to Laugier-Hunziker syndrome.
- Describe the management options for Laugier-Hunziker syndrome.
- Explain the importance of improving care coordination amongst interprofessional team members to improve outcomes for patients affected by Laugier-Hunziker syndrome.
Introduction
Laugier-Hunziker syndrome, also known as Laugier-Gerbig-Hunziker syndrome or Laugier-Hunziker-Baran syndrome or idiopathic lenticular mucocutaneous pigmentation, is a hereditary pigmentary disorder characterized by a unique expression of pigmentation over the mucosal, nail, and acral sites.[1][2]
The condition is known to be benign; nevertheless, a few associations with esophageal melanocytosis, actinic lichen planus, hypocellular bone marrow, and thrombocytopenia have been reported.[3]
Due to its close semblance to more serious conditions such as Addison disease, Peutz-Jeghers syndrome, Cronkhite-Canada syndrome, and lentiginosis profusa, this is usually classed as a diagnosis of exclusion.
Etiology
The most plausible mechanism for this syndrome is the presence of altered melanocytes in the epidermis.[4] The description is that of L-3,4 dihydroxyphenylalanine reactive melanocytes seen as large dendritic melanocytes. These cells are then capable of increasing melanogenesis.
Epidemiology
Laugier-Hunziker syndrome has more frequently been reported in the Asian population and displays a higher incidence in the Chinese population.[5][6] Cases have also been reported in European regions such as France and Italy. Based on gender predilection, a significant female preponderance has been described. Familial cases usually follow autosomal dominant as well as recessive traits, while sporadic cases are not uncommon.[6][7]
Histopathology
Histopathology of lesions shows increased pigmentation in the basal layer with a few dermal melanophages. Electron microscopy reveals multiple mature melanosomes within keratinocytes and melanophages.[4]
History and Physical
There have been juvenile and adult cases that carry specific disease presentation features but show increased severity in adult form.
Juvenile cases have been reported between the ages of 10 and 22 years, while most adult cases are seen between 43 and 55 years of age. The unique features are better described according to the sites affected.
Mucosal involvement is characterized by well-defined light brown to brown-black macules usually of size 0.1 to 0.5 cm over the oral and genital mucosa. Oral lesions are present over labial mucosa, buccal mucosa, hard palate, tongue, and posterior pharyngeal mucosa while genital lesions are seen on the glans and shaft of the penis in males and vulva in females. A few cases of isolated tongue pigmentation have been reported.
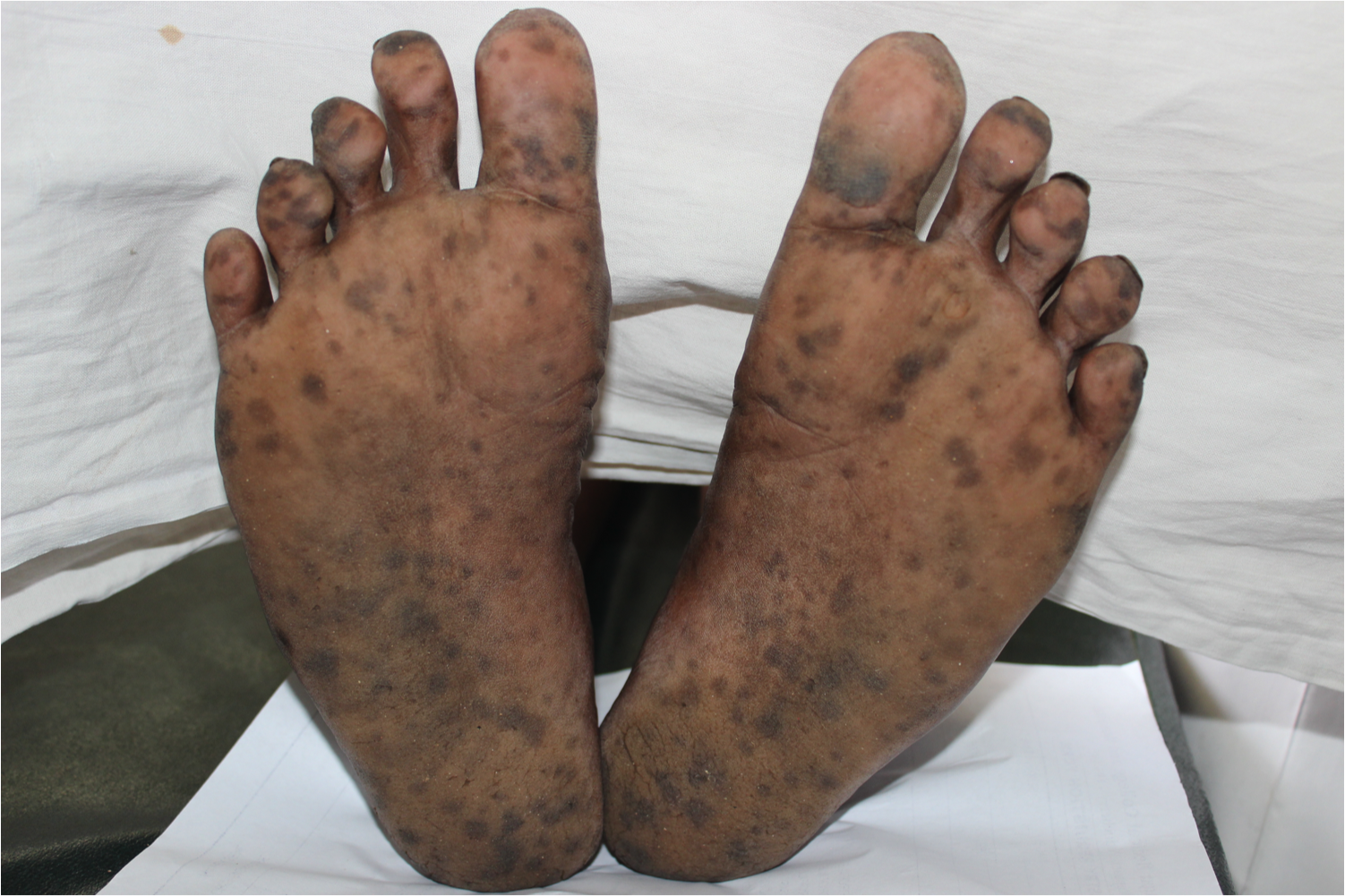
Acral or cutaneous involvement manifests as sharply marginated light brown to black lenticular macules and patches approximately 0.5 to 1.5 cm in size, specifically seen over distal two-thirds of all digits of the upper limb and bilateral plantar surfaces. Besides this, the lesions extend dorsally by involving the medial and lateral borders of digits.[2][8]
Nail involvement is seen in two-thirds of cases and can be divided into 4 types based on the extent of pigmentation.
- Single 1 to 2 mm longitudinal streaks
- Double 2 to 3 mm longitudinal streaks on the lateral parts
- Homogenous pigmentation involving radial or ulnar half
- Complete pigmentation
However, one or all types may be seen in the same patient. One striking feature of nail involvement is nail fold pigmentation termed as pseudo-Hutchinson’s sign.[9]
Rare reports of varied pigmentation include isolated tongue pigmentation, conjunctival pigmentation, neck and trunk pigmentation, and diffuse pigmentation are seen while the common finding of oral and acral involvements are more commonly encountered.[10][9][3][11][12]
Evaluation
With Laugier-Hunziker syndrome, owing to the insidious, although asymptomatic nature of the condition, there is delayed attendance years after onset is usually seen. A thorough history and clinical examination for signs of fatigue, weight loss, gastrointestinal (GI) involvement, and drug intake are necessary.
In the context of the exclusion of other disorders, investigations for the following are usually carried out after a complete physical examination and routine investigations.[5]
- Corticotrophin/adrenocorticotrophic hormone
- Serum cortisol
- Electrolytes
- Liver function tests
- Endoscopy
- Colonoscopy
- Ultrasound
- Thyroid function test
- HIV testing
- Radiographic barium studies
Recent reports of associated malignancies have suggested cancer screening, particularly in adult cases.[13]
Treatment / Management
The goal of therapy is purely cosmetic in cases of Laugier-Hunziker syndrome.
Treatment options include cryotherapy, Q-switched Nd:YAG laser, Q-switched alexandrite laser, erbium:YAG laser, CO2 laser, and diode laser.[14][15][16]
Differential Diagnosis
The differential diagnosis and their differentiating features include:
- Addison’s disease: Described as primary or secondary results from inadequate levels of adrenocorticotrophic hormone. Primary Addison disease results in hyperpigmentation described as more generalized with a predilection for sun-exposed areas and recent scars. A few reports of coexistent vitiligo have been described. Cutaneous lesions seem to precede systemic features of fatigue, lethargy, myalgia, nausea, personality changes, and hypotension.
- Peutz-Jeghers syndrome: An autosomal dominant condition characterized by intestinal polyposis and increased susceptibility to malignancies. Mucosal pigmentation differs from Laugier-Hunziker syndrome by crossing the vermilion border. Nail pigmentation is not seen in Peutz-Jeghers syndrome.
- McCune-Albright syndrome: Manifests with café-au-lait macules and not lentiginous lesions, as seen in Laugier-Hunziker syndrome. Other features are polyostotic fibrous dysplasia and precocious puberty.
- Cronkhite-Canada syndrome: This sporadic disorder manifests with gastrointestinal polyposis, anosmia, and dysgeusia. Hyperpigmentation is described with more proximal involvement (arms, legs) than in Laugier-Hunziker syndrome.
- Lentiginosis profusa and Leopard syndrome: Autosomal dominant syndrome characterized by multiple lentigines, hypertelorism, deafness, and cardiac conduction defects.
- Carney syndrome: An autosomal dominant syndrome comprising of lentiginous pigmentation, endocrinopathy, and malignancies.
- Bandler syndrome: Hyperpigmentation resembles Laugier-Hunziker syndrome; however, systemic involvement is seen as intestinal vascular malformations.
- Acquired immunodeficiency syndrome (AIDS): A diffuse hyperpigmentation may develop in advanced cases.
Other disorders include lichen planus, Smoker’s melanosis, Benign racial pigmentation, melanonychia striata, post-inflammatory hyperpigmentation, Nutritional deficiency (vitamin B12 and folate), and heavy metal poisoning (lead, arsenic, mercury, gold, bismuth, and silver).[17][18][19]
Generalized hypermelanosis can be seen with minocycline, phenothiazine, antimalarials, zidovudine, amiodarone, oral contraceptives, clofazimine, and chemotherapeutic agents.
Disorders that display pseudo-Hutchinson sign include Peutz-Jeghers syndrome, subungual hematoma, Bowen disease, and AIDS, while the true Hutchinson sign is specific to melanoma.
Prognosis
The pigmentary lesions of Laugier-Hunziker syndrome usually respond poorly to therapy, and it displays high recurrence. Of particular note of is sun avoidance following successful therapy, which has demonstrated lower rates of recurrence.[14]
Consultations
Reports of esophageal melanocytosis, actinic lichen planus, hypocellular bone marrow, and thrombocytopenia have been reported; however, these disorders are more likely coincident findings and not directly related to Laugier-Hunziker syndrome.[3] A recent report of pancreatic malignancy warrants evaluation in suspected cases.[13]
Pearls and Other Issues
- Laugier-Hunziker syndrome is a benign pigmentary condition that can be familial or sporadic.
- The absence of systemic features has usually led it to be named Laugier-Hunziker pigmentation.
- Classically presents as lenticular macules involving oral mucosa and palmoplantar skin.
- Nail pigmentation with pseudo-Hutchinson sign is a common finding.
- Lesions are usually resistant to treatment and display high rates of recurrence.
Enhancing Healthcare Team Outcomes
Laugier-Hunziker syndrome is a rare disease, and diagnosis may be challenging. A coordinated team approach between primary care nurse practitioners, physician assistants, physicians, and dermatologists is necessary to provide the best care of patients with this condition. [Level 5]