Continuing Education Activity
Optic pathway gliomas are benign CNS tumors that primarily affect children. Although typically slow growing, the location of these tumors makes resection impossible without loss of vision in at least one eye. This activity reviews the evaluation and management of patients with optic pathway gliomas. There is particular emphasis placed on the currently evolving treatment options and recommendations, which highlights the role of an interprofessional team in managing patients with this condition.
Objectives:
- Describe the pathophysiology of optic pathway gliomas.
- Outline the presenting clinical signs and symptoms of optic pathways gliomas.
- Review the current management options available for optic pathway gliomas.
- Describe the importance of collaboration and communication among the interprofessional team to ensure all treatment options for optic pathway gliomas are considered.
Introduction
Primary tumors of the optic nerve are relatively rare. They include the following:
- Optic nerve glioma
- Malignant optic nerve glioma
- Optic nerve sheath meningioma
- Ganglioglioma
- Primary lymphoma
The last two (gangliogliomas and primary lymphoma of the optic nerve) are very rare. Optic nerve gliomas and optic nerve sheath meningiomas make up just under 4% of orbital tumors. Optic nerve gliomas and meningiomas continue to attract controversy as their natural course is still being established. Treatments for both of these conditions continue to evolve.
Optic nerve gliomas are benign tumors classified as pilocytic astrocytomas. They make up half of all primary optic nerve tumors and between 1.5 and 4% of all orbital tumors.
Etiology
Optic pathway gliomas (OPG) are low-grade neoplasms arising from the pre-cortical optic pathways. OPG can involve the optic nerve, optic chiasm, optic tracts, optic radiations, or the hypothalamus. These neoplasms may arise sporadically or in association with neurofibromatosis type 1 (NF1).[1] When in association with NF1, neurofibromin, a tumor suppressor on chromosome 17q, is inactivated. This turns on the RAS signaling pathways, resulting in RAS-induced tumors.[2] When arising sporadically, the most common genetic alteration identified is a BRAF-KIAA1549 fusion.[3]
Several studies have shown that the prognosis of optic pathway gliomas in patients with NF1 is better with a better visual prognosis.[4] This subset of gliomas present later and progress over a longer time when compared to spontaneous gliomas. All patients with gliomas need ophthalmic follow-up with vision assessment. Electrophysiologic assessment may be needed in the very young, where vision can be difficult to assess.
Epidemiology
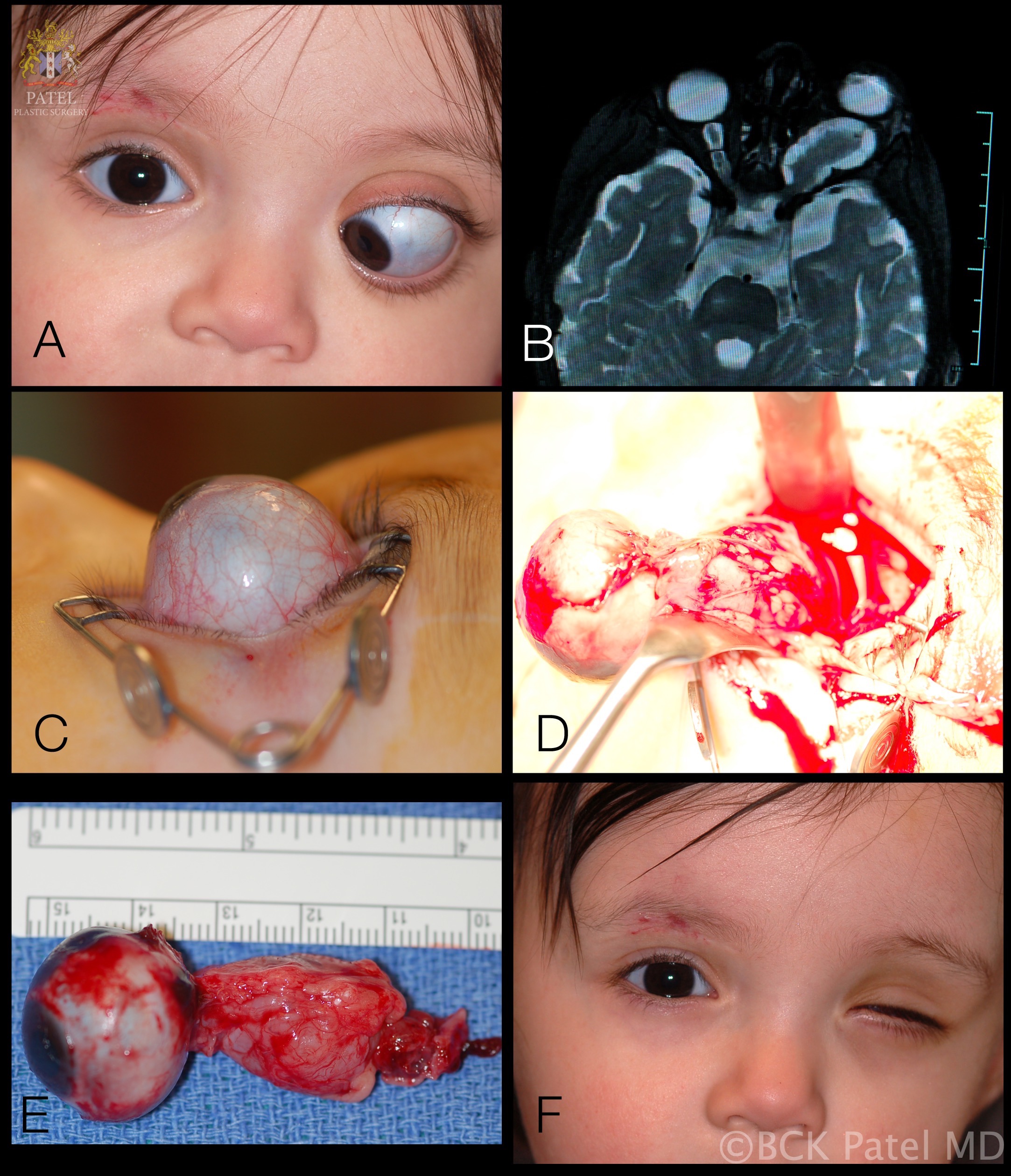
Optic pathway gliomas most commonly affect children under ten years old and account for 3 to 5% of childhood central nervous system tumors.[2] However, gliomas have been found in patients ranging from birth to 79 years of age. 71% of cases are found in the first decade of life and 90% within the first two decades. When all optic gliomas are considered, the mean age at presentation is 8.8 years. (fig. 1, fig. 2)
Approximately 15% to 20% of patients with neurofibromatosis (NF) 1 eventually develop an OPG, but only 30% to 50% of those patients will be symptomatic.[1][5] The incidence of NF1 in patients presenting with optic nerve gliomas ranges from 10% to 70% (with an overall incidence of 29%).[6] Males and females were similarly affected.[7][1]
Optic pathway gliomas occur equally in males and females. However, with gliomas confined to the optic nerve, there is an increased incidence in females (65%) compared to males (35%). When tumors involve the optic chiasm, males and females are equally affected.
Gliomas at presentation are confined to the optic nerve alone in 25% of cases, but this leaves 75% of cases that show chiasm involvement.[8][9] When the chiasm is involved, 40% of patients will develop an extension of the tumor to the hypothalamus or third ventricle.
Histopathology
Gliomas were previously regarded to be benign hamartomas with limited growth. However, we now know that the rate of growth is variable with a rare occurrence of malignant change. Furthermore, the tumors have a tendency to invade the leptomeninges. Therefore, gliomas are now regarded as true neoplasms with the ability to invade locally. Gliomas arise from astrocytes of the optic nerve and visual pathway. These proliferating astrocytes extend through the pia mater and into the arachnoid and subarachnoid space, where fibrovascular and meningeal cell proliferation, which is reactive, may occur. This has been termed arachnoidal hyperplasia. When exuberant, there will be an appearance of tumor growth. Tumor enlargement is caused by proliferation of the neoplastic cells by the reactive arachnoidal cell proliferation or by the accumulation of mucosubstance, which is secreted by astrocytes and is PAS-positive.
- Usually, WHO grade I pilocytic astrocytoma with immature astrocytes
- Tumor cells have spindle-shaped nuclei and appear pilocytic or hair-like
- Biphasic pattern with varying proportions of piloid areas alternating with spongy areas
- Intracytoplasmic Rosenthal fibers and eosinophilic granular bodies
- Microcystic areas
- Focal calcification may also be seen
- Rosenthal fibers (tapered corkscrew-shaped, brightly eosinophilic, hyaline masses[10]
History and Physical
The location of the tumor determines the presenting symptoms and signs.
Loss of Vision
Children will often not complain of loss of vision. Overall, 85% of patients with glioma will lose some vision, and over time, approximately 25% will retain vision between 20/20 and 20/40. About 60% of patients will develop vision worse than 20/300. Patients may have an afferent pupillary defect, and where visual field assessment is possible, there may be visual field defects. Optic nerve atrophy (seen in 60% of patients upon fundoscopic examination) and optic disc edema (seen in 50% of intraorbital tumors) may be seen. Patients with chiasmal tumors will show optic nerve edema in 20% of cases, and these will have associated orbital optic nerve involvement.
Proptosis
Proptosis is seen in 95% of patients presenting with glioma is the commonest presenting sign. It is commonly seen with optic nerve gliomas but less so when the tumor is confined to the optic chiasm (seen in less than 20% of patients). When proptosis is seen in patients with optic chiasm glioma, there is almost always concomitant involvement of the optic nerve.
Limitation of ocular motility: seen infrequently with optic nerve gliomas. Limitation of ocular motility may be seen in 30% of optic nerve gliomas confined to the orbit and 20% of patients where the chiasm is involved.
Headache/Pain
The most common presenting symptom of OPG is headache, although it is only found in about 30% of patients, mostly in chiasmal involvement.
Other symptoms and signs include nystagmus, spasmus nutans, convulsions, nausea, dizziness, strabismus, developmental regression, and growth retardation. Hydrocephalus may develop when the tumor spreads from the chiasm. These patients may also develop hypothalamic-pituitary dysfunction with precocious puberty, growth hormone deficiency, and deficiency of gonadotropin, TSH, and ACTH.
In summary, the optic pathway gliomas present with:
- Early vision loss 88%
- Optic disc swelling 35%
- Optic nerve head atrophy 59%
- Proptosis in orbital tumors 94%
- Proptosis in chiasmal tumors 22%
- Nystagmus 24%
- Hypothalamic signs 26%
- Increased intracranial pressure 27%
Evaluation
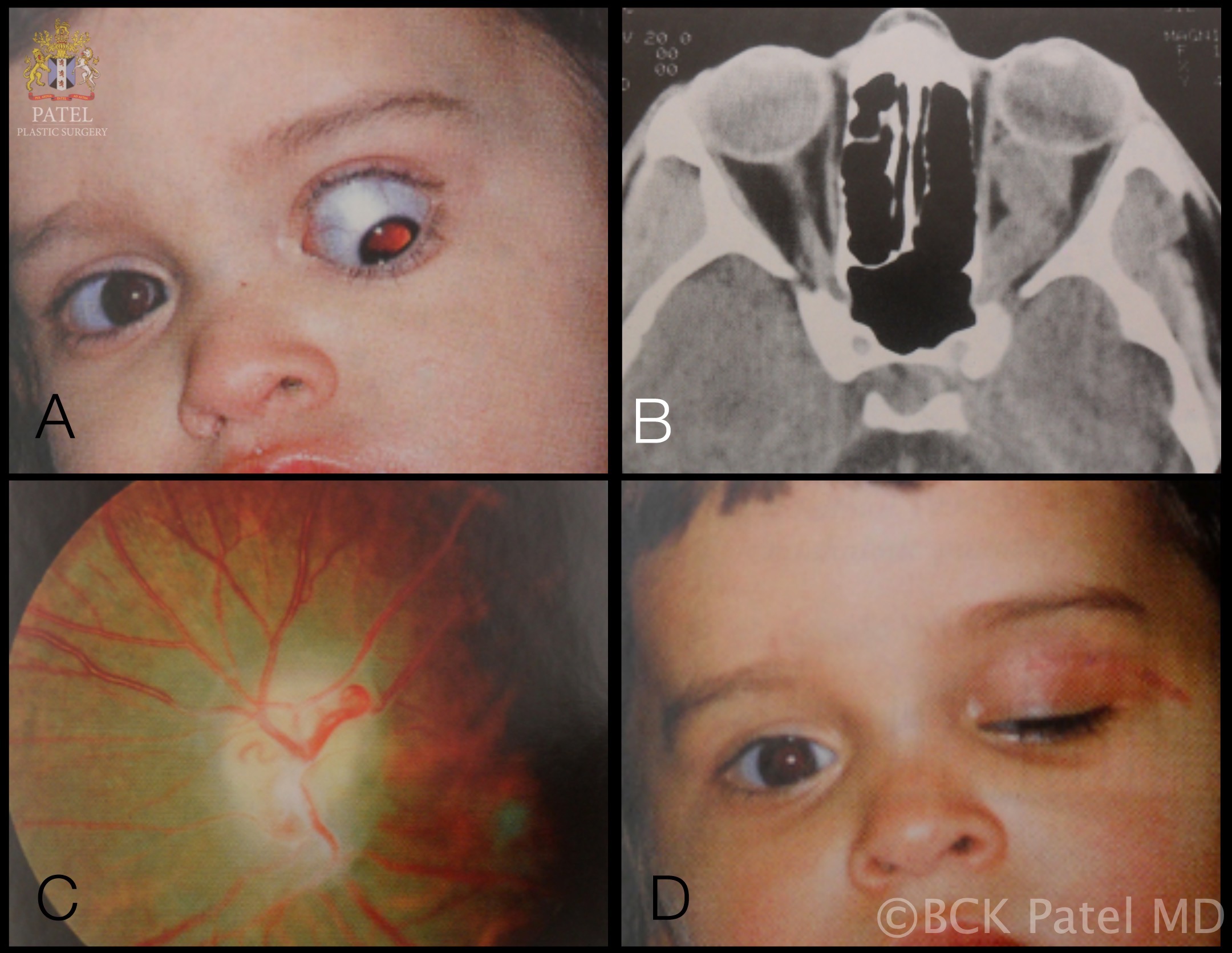
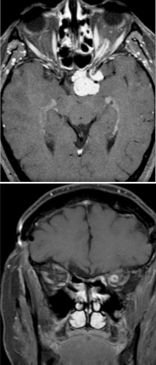
Work-up initially includes magnetic resonance imaging (MRI) of the brain and orbit. Boire et al. found that about 80% of patients have radiographical progression on MRI.[1] Enlargement of the optic canal will be seen in 80% of patients where the optic nerve is involved. In 25% of patients with chiasmal glioma, sella turcica enlargement and J-shaped excavation may be found. When evaluating chiasmal, hypothalamic, and optic tract tumors, MRI is superior to computed tomography (CT).
On MRI, gliomas will demonstrate slightly prolonged T1 relaxation times, and the image of the tumor will be isointense or slightly hypointense compared to the normal optic nerve. T2-weighted images will show a hyperintense image with prolonged T2 relaxation time.
CT will show enlargement of the optic nerve and/or chiasm. When contrast is used, the enhancement will be mild to moderate and overall less than that seen with optic nerve sheath meningiomas. The optic nerve will show a fusiform swelling, but rounded and exophytic changes may also be seen. Calcification in optic pathway gliomas is uncommon but may be seen.
Genetic testing for neurofibromatosis is always warranted. A biopsy is typically unnecessary because the diagnosis can be made based on imaging and clinical examination. However, new molecularly targeted therapies may make genetic testing of the neoplasm increasingly useful.
Distinguishing Optic Nerve Gliomas and Meningiomas on CT and MRI Scans
Both optic nerve gliomas and meningiomas can show a diffuse enlargement of the optic nerve, and both may also show a globular or fusiform enlargement of the optic nerve. However, meningiomas will show "tram-tracks," which are caused by the thickened and denser optic nerve sheath in meningiomas (Fig 3), resulting in a central lucency, which is the residual optic nerve. Calcification is seen in 20 to 50% of meningiomas but very rarely in gliomas. There is an increased signal intensity with meningiomas compared to the normal nerve on MRI on both T1- and T2-weighted sequences. Optic nerve gliomas will show an isointense or slightly hypointense signal on T1 but hyperintense on T2 sequences.
Treatment / Management
Natural History
The natural history and course of these gliomas need to be appreciated so that the proper management is undertaken. Gliomas grow slowly but have the potential to spread into the surrounding tissues, including around the optic nerve, the chiasm, and into the adjacent brain. In most patients, with slow growth, there is stabilization of the vision. In about 40% of patients, however, continued growth may be seen.
Management
Treatment is managed by an interdisciplinary team of neuro-ophthalmologists, hematology-oncologists, oculoplastic surgeons, and radiation-oncologists, depending on the case. Management is tailored based upon multiple factors, including symptoms, nature of the tumor, tumor progression, vision and vision change, clinical course, NF1 status, neoplasm genetics, patient age, and patient preference. Observation is the first choice until significant visual deficits or radiographical progression is seen.
The management of gliomas is summarized a follows:
Optic Nerve Glioma
- If stable, simply observe clinically and radiologically
- In the presence of vision loss and severe proptosis, surgery may be considered
- If there is posterior progression of the tumor with the risk of invasion of the chiasm or surrounding tissues, surgical resection can be undertaken
Optic Chiasm Glioma
- If stable, observe with sequential radiological assessment
- If there is progression in patients of less than 10 years of age, consider chemotherapy.
- If there is progression in patients more than 10 years of age, combine chemotherapy and radiotherapy.
Midbrain Glioma
- If stable in patients for more than 10 years, consider chemotherapy and radiotherapy
- If stable in patients less than 10 years, consider chemotherapy
- If there is progression, a combination of chemotherapy and radiotherapy is considered.
Observation
In 80% of patients, there is an initial phase of visual deterioration followed by stabilization. However, stable vision does not always equate with a stable tumor, as tumor progression may be seen even in these patients. Long-term survival has shown a good prognosis when patients are followed conservatively with no intervention.[11][12]
Surgery
Whereas surgical resection was considered the treatment of choice until recently, it is now recognized that surgery should be limited to obtaining a biopsy for diagnostic purposes, or to resect tumors when they cause excessive proptosis and pain or when they extend posteriorly, threatening involvement of the optic chiasm. It is now known that optic nerve glioma will rarely progress to the chiasm or damage the contralateral fibers.[13] Certainly, when vision is lost, surgical resection helps to reduce the proptosis and helps with orbital pain. We generally remove these tumors via the lateral trans-orbital approach with preservation, whenever possible, of the globe and all the extraocular muscles as this gives an excellent cosmetic result. It should be noted that if surgery is performed when vision is present, the risk of loss of vision is substantial.
Radiotherapy
Radiotherapy has continued to be controversial. Although initial studies showed no benefit on survival or vision preservation, more recently, it has been shown that stabilization of vision and more limited progression of the tumor may be achieved.[14][15] Radiation therapy, whether by external beam radiation, stereotactic radiosurgery, or proton beam therapy, is a potential option for refractory cases. Because of the risk for disfigurement and other radiation-induced side effects, this is typically reserved for older children and teens. Side-effects to the central nervous system in these children may also occur.
Chemotherapy
In most cases, chemotherapy with vincristine/carboplatin is considered first-line treatment. It has about a 70% progression-free survival (PFS) and has no increased risk of secondary malignancy or treatment-related mortality. The primary adverse event is hypersensitivity reactions to carboplatin, seen in 40% of patients. Other chemotherapy regimens include Thioguanine/procarbazine/CCNU/vincristine (TPCV) and cisplatin/etoposide, and temozolomide. These have similar PFS but have an increased risk of secondary leukemia and should, therefore, be avoided in patients with NF1.[13]
Bevacizumab, a VEGF inhibitor, has also been shown to be effective in stopping or reversing the growth of OPG. It has been reportedly used as both monotherapy and combined therapy with vinblastine or irinotecan. Hwang et al. reported 14 cases of refractory OPG that all remained stable or regressed during treatment. However, 4 of those patients had tumor progression after discontinuation of therapy. The most common adverse events involved gastrointestinal toxicity, with most resolving with treatment cessation.[16]
Recently, gene-targeted therapy has become increasingly common. This has increased the utility of tumor biopsy. The most common genetic aberration is a BRAF-KIAA1549 fusion. This activates the MEK/MAPK/ERK pathway causing proliferation, survival, and tumorigenesis. MEK inhibitors target this pathway and report a 69% 2-year PFS for refractory OPG.[17] The most widely used MEK inhibitor currently is trametinib, but over a dozen alternatives are currently undergoing clinical trials.[18] The most common side effects of MEK inhibitors involve dermatologic conditions; however, vision-threatening ocular adverse events including optic neuropathy, retinal vein occlusion, uveitis, neurosensory retinal detachment, and MEK inhibitor-associated retinopathy have been reported. Fortunately, these are rare and typically reversible in children.[13]
Subtypes of Gliomas
1. Gliomas confined to the optic nerve
When these tumors, which are confined to the optic nerve, are treated conservatively or incompletely resected, recurrence or progression is seen in 17%.[6] In this group, there is a 12% mortality from intracranial extension. Overall, the prognosis for vision is very good, with more than 90% remaining stable over years. If the optic nerve glioma is surgically resected with or without radiotherapy, the mortality rate drops to nearly zero, but, of course, there is loss of vision. If posterior extension of the glioma is observed, and there is loss of vision, it is reasonable to resect optic nerve gliomas to prevent progression but also reduce mortality.
2. Gliomas that extend to the chiasm
If there is no invasion of the adjacent third ventricle or the hypothalamus, these tumors behave like those confined to the orbital optic nerve. Left untreated, these chiasmal gliomas have a mortality of 17% over 10 years. If the tumor extends into the surrounding tissues (hypothalamus or third ventricle), then mortality rises. If the tumor is partially resected from the chiasm, there is a high rate of recurrence or progression (64%). Untreated, the visual prognosis is very good, with 80% remaining stable. It is thought that fractionated radiation may slow the growth of tumor in the chiasm without the risk of loss of vision that surgery entails. Overall, when all modalities of treatment are considered, chiasmal gliomas have a mortality of 22%, recurrence or progression of tumor 43%, and visual stabilization in 68%.[6]
3. Gliomas that extend to the chiasm and invade the hypothalamus
The mortality rate in patients where the chiasmal tumor extends into the hypothalamus or third ventricle is more than 50% over 15 years. Mortality may be reduced with radiotherapy and chemotherapy, but recurrence or progression is still seen in 52% of cases.[6]
Differential Diagnosis
Optic nerve gliomas can almost always be diagnosed by MRI imaging alone because of pathognomonic features. An optic nerve sheath meningiomas may have a similar location, but they typically have distinct radiographical features and are rare in children.
- Meningioma
- Rhabdomyosarcoma
- Orbital lymphoma
- Benign lymphoproliferative lesion
- neuroblastoma
- Epithelial tumor
- Inflammatory lesions
- Infectious lesion
- Metastasis of extraocular tumor
Medical Oncology
Malignant Optic Nerve Glioma
It is important to recognize a rarer type of glioma, which is seen in older patients. Since Hoyt et al. first described aggressive optic nerve glioma in adults which behaved in a malignant manner, there have been some 60 additional reported cases.[19][20] The age range of these tumors is 6 to 79 years, but most occur in middle age with a mean age presentation of 48 years.[19] Males are affected in 65% of cases. The main site of tumor occurrence is the optic chiasm and is almost always bilateral. Extension of the malignant tumor from the chiasm into the orbital optic nerve is seen in 23% of cases. In 50% of cases, the tumor extends posteriorly from the chiasm, along the optic tracts, into the hypothalamus or the temporal lobe.
Clinical Course
These patients present with a rapid loss of vision, first presenting on one side and then both. When first seen, these patients show vision deterioration in both eyes in 63% of cases. The condition may be misdiagnosed as anterior ischemic optic neuropathy or optic neuritis because of the presentation. Most patients will show optic disc edema, going on to develop optic atrophy if the patient survives. Unlike optic pathway gliomas, proptosis is only seen in 20% of patients.
Evaluation
In 80% of cases, CT or MRI will show optic chiasm enlargement and enhancement. Intracranial portions of the nerves will also show involvement.
Diagnosis
This is only possible with a biopsy and histopathology.
Histopathology
These are anaplastic astrocytomas (WHO grade III) or glioblastomas (WHO grade IV). Malignant astrocytes with pleomorphic nuclei, necrosis, and vascular proliferation are seen. Invasion into the surrounding brain is seen.
Treatment
Neither surgery nor radiotherapy has been found to change the prognosis of this condition. In a few patients, survival may be improved by a few months with an aggressive combination of radiotherapy, chemotherapy, and surgery.
Prognosis
All patients progress to complete vision loss within months of presenting. The mortality rate is 100%, with a mean survival of less than 1 year.
Prognosis
NF1 associated OPG is generally considered to have a better prognosis. However, this appears to be driven primarily by patient selection bias and tumor location. Many OPGs show little to no progression over time and may remain asymptomatic for the patient’s entire lifetime. These are often found incidentally in patients with NF1 but are almost always symptomatic in sporadic cases.[1][21] NF1 associated OPG is also significantly more likely to be pre-chiasmatic. Robert-Boire et al. found that progression was equally likely with NF1 and sporadic cases. However, out of 40 patients with OPG, the only patients with progression-associated mortality had bilateral chiasmatic or post-chiasmatic sporadic tumors.[1]
An excellent detailed review of the literature by Jonathan Dutton has shown that when all patients with optic pathway gliomas in all locations and with all forms of treatment (including observation) are considered, tumor recurrence or progression occurs in 38% of cases.[6] Tumor-related mortality with a mean follow-up of 11 years is 36%. Approximately 55% of patients retain stable vision or demonstrate improvement, with only 45% developing progressive vision loss.
Spontaneous regression of optic gliomas is rare but has been reported.[22] Malignant transformation of optic pathway gliomas is extremely rare but has also been reported.[23]
Complications
Surgical resection of an optic nerve glioma will invariably lead to blindness of the affected side. Resection of a chiasmal or post-chiasmal OPG will likely lead to bilateral visual deficits. Resection often entails enucleation of the affected eye. Radiation therapy can lead to significant facial disfigurement, particularly in younger children.
Enhancing Healthcare Team Outcomes
Management of gliomas requires an interprofessional team of neuro-ophthalmologists, hematology-oncologists, orbital and oculoplastic surgeons, radiation oncologists, and specialty trained nurses. An experienced electrophysiologist may be needed as vision assessment may be difficult or impossible in very young children. We always involve a clinical psychologist in the clinic visits and when any surgery or treatment is performed. We also introduce the family to our ocularist early and prior to any globe-removal surgery, which is sometimes needed. When vision is lost in one eye, we educate the family about monocular vision with the help of our psychologist. [Level 3] The medical professionals form an interprofessional team that improves communication and patient outcomes. [Level 5]