Continuing Education Activity
Orbital pseudotumor, also known as orbital inflammatory pseudotumor (OIP), idiopathic orbital inflammation (IOI), or idiopathic orbital inflammatory syndrome (IOIS), is a benign, noninfectious, nonneoplastic, space-occupying, inflammatory condition of the orbit and peri-orbit without identifiable local or systemic causes. This condition can affect any or all of the orbit's structures, including the lacrimal gland, extraocular muscles, optic nerve, and surrounding soft tissues. Despite its name suggesting a tumor-like appearance, orbital pseudotumor is characterized by an inflammatory reaction rather than neoplastic features. The exact cause of orbital pseudotumor is poorly understood, but it is thought to involve an abnormal immune response or inflammatory process.
This activity provides an in-depth review of current theories on the etiology, clinical manifestations, and pathogenesis of orbital pseudotumor. It covers differential diagnosis, laboratory and imaging tests, and treatment options for managing this condition. Additionally, this activity discusses the roles of the interprofessional healthcare team in implementing best practices for diagnosing, treating, preventing complications, and managing patients with orbital pseudotumor to improve patient outcomes. By participating in this activity, healthcare professionals can enhance their diagnostic and therapeutic skills to better care for patients with this complex inflammatory condition of the orbit.
Objectives:
Identify the clinical manifestations of orbital pseudotumor.
Screen patients with orbital symptoms for signs of orbital pseudotumor, including a thorough history and physical examination.
Implement evidence-based treatment strategies for diagnosing and managing orbital pseudotumor.
Collaborate with the interprofessional team to implement personalized strategies for improving care coordination and communication, advancing the diagnosis and treatment of orbital pseudotumor, and improving outcomes.
Introduction
Orbital pseudotumor, also known as orbital inflammatory pseudotumors (OIP), idiopathic orbital inflammation (IOI), idiopathic orbital inflammatory syndrome (IOIS), idiopathic orbital inflammatory pseudotumor (IOIP), or nonspecific orbital inflammation (NSOI) is a benign, space-occupying, and noninfectious inflammatory condition of the orbit but may extend in the periorbital area.[1] No identifiable infectious, systemic, or neoplastic disorder is associated with it. It is the third most common orbital disease in adults, following thyroid orbitopathy and lymphoproliferative diseases.[2]
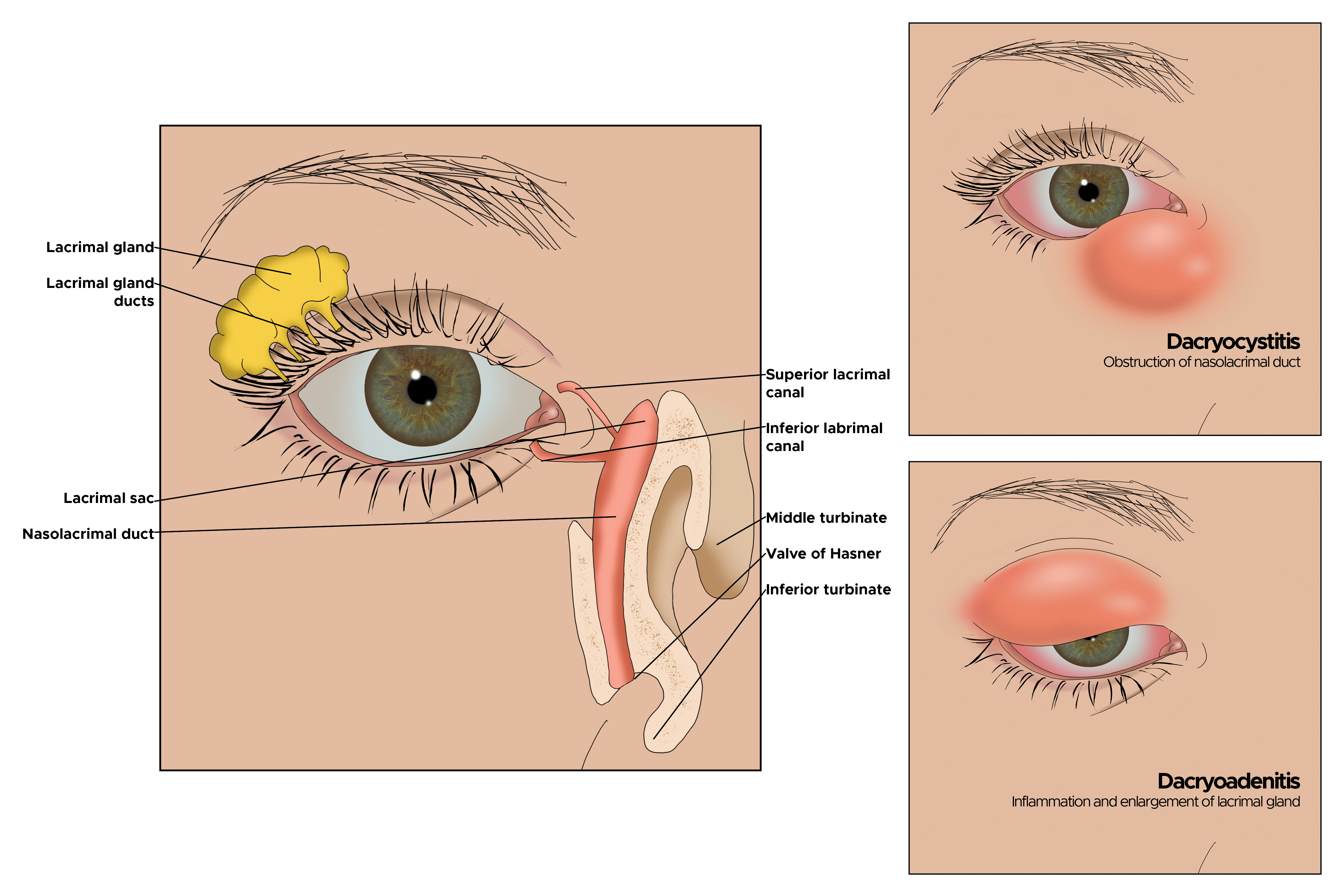
Many orbital inflammations may be associated with systemic conditions or remote organ dysfunction. Categories of orbital pseudotumor according to location include anterior, diffuse, posterior, or apical. Other classifications include myositis, dacryoadenitis, periscleritis, perineuritis, and focal mass.[3]
Orbital pseudotumor is rare in children. The most common ophthalmic findings include periorbital edema and blepharoptosis. A palpable mass may be present. On orbital radiography, common findings are dacryoadenitis, orbital mass, or myositis.[4] In children, systemic signs are present in up to 50% of patients. Headache, emesis, anorexia, lethargy, and fever are the most common systemic signs. Additionally, there are likely associations with iritis, uveitis, disc edema, and peripheral eosinophilia.[5]
The pathophysiology of orbital pseudotumor, first identified more than a century ago,[6] is still unknown, adding to the difficulties in diagnosis and uncertainty surrounding treatment. However, advances in imaging, histology, and immunology have illuminated the underlying mechanisms of the illness, previously believed to be essentially idiopathic.[7] These mechanisms involve a complex interaction of immunological dysregulation, viral triggers, and genetic predisposition.[8][9]
Understanding the clinical spectrum and diagnostic criteria of orbital pseudotumor is essential for accurate diagnosis and effective treatment. Patients commonly present with unilateral proptosis, discomfort, diplopia, and visual abnormalities, which can be either acute or subacute.[10] These symptoms often resemble other orbital diseases such as neoplasms, infections, or thyroid eye disease.[11] Clinical assessment, imaging modalities (such as computed tomography [CT] and magnetic resonance imaging [MRI]), and occasionally histological examination acquired via biopsy are utilized to differentiate orbital pseudotumor from these conditions.[12][13]
Etiology
The term "orbital pseudotumor" encompasses various inflammatory disorders affecting the orbit with diverse etiological causes. Despite extensive research efforts, the exact etiology of orbital pseudotumor remains unknown. However, it is likely the result of a complex interaction between genetic predisposition, environmental stimuli, and dysregulated immune responses.
There has long been a suggestion that infectious agents could trigger orbital pseudotumor. Several microbiological agents, including viruses, bacteria, and atypical species, have been associated with increased orbital inflammation, although no single pathogen has been consistently identified. Recent research has indicated a possible link between the emergence of orbital pseudotumor and certain viral infections, including herpesviruses (HSV),[14] SARS-CoV-2[15], and Epstein-Barr virus (EBV).[16]
In at-risk patients, HIV can cause orbital myositis, leading to a T-cell-mediated proinflammatory orbital response.[17] Upper respiratory tract infections, sinusitis, and dacryoadenitis have been reported to be associated with orbital pseudotumor.[18][19] Among infections, the presence of Streptococcal pharyngitis or viral upper respiratory infection has reportedly correlated with the occurrence of orbital pseudotumor. Molecular mimicry of the foreign antigen with self-antigens is one theory that explains IOI after infection.[3]
Various rheumatologic disorders, systemic diseases, and autoimmune pathologies such as granulomatosis with polyangiitis (formerly called Wegener granulomatosis), thyroid eye disease,[20] giant cell arteritis,[21] systemic lupus erythematosus (SLE), discoid lupus,[22] Behcet disease,[23] sarcoidosis, Churg-Stauss syndrome,[24] rheumatoid arthritis, polyarteritis nodosa, multifocal fibrosclerosis (now categorized as IgG4-related disease), Crohn disease,[25] psoriasis, and ulcerative colitis have been reported to be associated with orbital inflammation.[5][26] These conditions can present diverse clinical manifestations, complicating the diagnostic process.
Several theories have been proposed to explain the inflammation that causes orbital pseudotumor, including immune dysregulation.[27] The pathophysiology of orbital pseudotumor has been reported to be driven by immunological processes, with orbital inflammation linked to the initiation and persistence of dysregulated innate and adaptive immune responses.
Numerous immune cells, such as T lymphocytes, B lymphocytes, macrophages, eosinophil granulocytes, neutrophils, and dendritic cells, have been implicated in the inflammatory cascade inside the orbit.[28] Reports exist of high levels of inflammatory cytokines such as interleukins, interferon (IFN), and tumor necrosis factor (TNF) in histopathological specimens and increased expression of CD20 and CD25.[3] Gene expression profiling methods have recently shown upregulation involving immunoglobulin and receptors but with downregulation of alcohol dehydrogenase 1B, adiponectin, leptin receptor, and C1Q.[3]
Environmental factors have been suggested to be linked with increased orbital inflammation, even if the precise mechanisms by which environmental variables contribute to the development of orbital pseudotumor are still unknown. Studies have reported increased odds ratios of orbital inflammation in obese patients with high body-mass index (BMI) values.[29] It is hypothesized that low-grade chronic inflammation due to an abundance of immune cells in adipose, seen in obese patients, can trigger immune imbalance and favor orbital inflammation.[30]
Certain medications such as lithium, bisphosphonates, and chemotherapy have been reported to affect inflammatory responses, leading to inflammatory mediator release, increased cytokine production, and T-cell activation, which could help trigger orbital inflammation.[31][32]
Epidemiology
Orbital pseudotumor accounts for approximately 8% to 11% of all orbital disorders.[2][33] Studies have reported an incidence of orbital pseudotumors of 6% to 16% in patients with suspect orbital tumors.[4] A retrospective study in an eye hospital in India, based on over 6000 patients with orbital disease in 10 years, showed that 23% of patients had orbital pseudotumor.[34] A study from Tokyo regarding 1000 patients with primary orbital tumors from 1995 to 2019 reported that 27% of patients had benign orbital tumors.[35]
Orbital pseudotumor is more prevalent in adults, especially middle-aged females, and has been reported worldwide among all ethnic groups. The most common site of involvement is the lacrimal gland.[1] The mean age of presentation is typically between the third and sixth decades of life, with bilateral cases being rare in adults.[4] However, bilaterality is more common in children. The recurrence rate in the pediatric population is as high as 76%, and its rate may or may not be associated with bilaterality.[7] The reported recurrence rate of NSOI following resolution is from 33% to 58% of patients.[36]
Histopathology
The necessity of an orbital biopsy is up for debate since typical IOI cases can often be effectively treated with systemic corticosteroids without it.[3][37][38] Reports suggest that biopsies should be considered only if there is no or incomplete response to steroids.[38] Patients with a history of systemic malignancy or persistent diagnostic uncertainty should undergo a biopsy.[39]
Histopathological evaluation is crucial in the diagnosis and characterization of orbital pseudotumor.[30] Histopathologists can determine distinctive histological characteristics that help differentiate orbital pseudotumor from other ocular diseases and provide insights into its underlying pathophysiology by analyzing tissue samples obtained through biopsy or surgical excision. Studies have reported histopathological findings related to orbital pseudotumor, shedding light on the complex inflammatory nature of the condition and assisting in the development of more accurate diagnostic and treatment planning techniques.[40]
Histopathologic examination through fine-needle aspiration or incisional/excisional biopsy is the gold standard for definitive diagnosis.[41] Biopsies are preferred for palpable orbital lesions, like those of the lacrimal gland or superficial inflammations,[42] considering that deep orbital lesions and myositis biopsies can damage the optic nerve.[43] However, a fine needle biopsy may have a low yield due to the firm nature of the tumor. Refinement of immune staining has increased specificity and sensitivity in differentiating lymphoma from pseudotumor.
Research has described 5 patterns of acute inflammatory pseudotumors: anterior and diffuse acute pseudotumors, anterior or diffuse orbital infiltration, lacrimal involvement, posterior or apical involvement, and myositic infiltration.[1] These patterns help in understanding the varied clinical presentations and guiding appropriate diagnostic and therapeutic approaches.
On histopathology, there is a pleomorphic inflammatory cellular response consisting of lymphocytes, plasma cells, macrophages, polymorphonuclear leukocytes, and eosinophils.[1][3] Fibrovascular tissue reaction is common, and when extensive, it leads to a diagnosis of sclerosing orbital pseudotumor. Other features include myositis, perineuritis, periscleritis, and sometimes, a lacrimal gland mass may be present. Granulomatous inflammation is uncommon, but a granulomatous inflammatory variant mimics sarcoidosis. Histiocytic infiltration and well-formed noncaseating granulomas may be present.
Histopathological examination of orbital pseudotumor usually reveals a dense inflammatory infiltrate of lymphocytes, plasma cells, macrophages, and occasionally eosinophils. The perivascular distribution of the inflammatory infiltrates encircles blood vessels and expands into the surrounding fibrous tissue. The prevalence of T lymphocytes, especially CD4+ helper T cells, among the inflammatory infiltrates has been shown by immunohistochemical investigations, indicating a potential involvement for cell-mediated immune responses in the pathophysiology of orbital pseudotumor.[3][30]
Common histological findings seen in orbital pseudotumors include fibrosis and granulomatous reactions in addition to inflammatory infiltrates.[44] Fibrosis can cause architectural deformation and tissue remodeling inside the orbit due to the deposition of collagenous connective tissue. The granulomatous response is observed in several subtypes of orbital pseudotumor, including granulomatous orbital inflammation,[45] characterized by the production of epithelioid histiocytes and multinucleated large cells. These histological findings reflect the chronicity and complexity of the inflammatory process in orbital pseudotumor.
Orbital pseudotumor may exhibit vascular alterations, such as vasculitis, endothelial cell growth, and perivascular hyalinization.[46] These vascular changes contribute to the edema, ischemia, and tissue destruction observed in affected orbital tissues. Immunohistochemical investigations have demonstrated increased expression of VEGF and other angiogenic factors within the inflamed orbit, suggesting a potential role for angiogenesis in the pathophysiology of orbital pseudotumor.
By evaluating the expression of specific markers of inflammation, proliferation, and immune activation, immunohistochemical investigation of orbital pseudotumor tissues might offer further diagnostic and prognostic information. Numerous immunohistochemical and molecular markers, including CD3, CD20, CD68, Ki-67, and cytokines like interleukin-6 (IL-6) and tumor necrosis factor-α (TNF-α), have been linked to orbital pseudotumor in recent research. These markers can help with subtype categorization, disease activity assessment, and therapy response prediction in patients with orbital pseudotumor.[47]
The presence of granulomatous inflammation, vasculitis, or necrosis generally excludes the diagnosis of IOI.[28] The immunohistochemical assessment of plasma cells for IgG4 positivity is required to exclude IgG4-RD. Tissue plasma cell IgG4 positivity is not a prominent finding in IOI.[48]
History and Physical
Due to its varied and sometimes vague presentations, orbital pseudotumor poses a clinical challenge. A thorough approach to history-taking, physical examination, and identification of typical signs and symptoms is required. The clinical appearance might vary greatly depending on the orbital components affected and the degree of inflammation.
Orbital pseudotumor is diagnosed by excluding other orbital diseases with similar presentations. Thus, systemic diseases such as sarcoidosis, granulomatosis with polyangiitis, Sjogren syndrome, IgG4-related disease (IgG4-RD), lymphoproliferative and histiocytic disorder, xanthogranulomatous disease, or metastatic disease may be elicited through a careful history-taking and a review of previous medical conditions.[28] A negative history may necessitate further laboratory testing to rule out conditions that orbital pseudotumor may mimic.
History
Understanding the onset, duration, progression, and related systemic symptoms of orbital pseudotumor requires a thorough history. Physicians should inquire about the time of day symptoms occur and how long they last. Patients often report the subacute onset of symptoms, developing over a few days to weeks, although some instances may present more subtly or acutely.
The clinician should assess for soreness, redness, swelling, or discomfort around the eyes. Additionally, patients may experience diminished visual acuity, diplopia (double vision), or blurring. While orbital pseudotumor primarily affects the orbit, fever, malaise, weight loss, or arthralgias are occasionally linked to systemic symptoms. Inquiring about recent viral infections or autoimmune diseases may provide important diagnostic clues. Patients with a history of autoimmune illnesses, such as thyroid eye disease, SLE, or granulomatosis with polyangiitis (GPA), may be at higher risk of orbital pseudotumor. Inquiring about recent orbital trauma, surgery, or radiation therapy is also essential.
Signs and Symptoms
IOI presents highly variable clinical features, ranging from a diffuse process to a specific focal target like the lacrimal gland and extraocular muscles.[49] The presentation can be acute, subacute, insidious, chronic, and relapsing.[3][50] Inflammatory symptoms such as redness and swelling are common, with periorbital erythema and edema being typical.
Acute proptosis, a forward displacement of the globe due to intraorbital tissue expansion or swelling, often prompts patients to seek emergency care.[9] Insidious cases may present with gradual proptosis, swollen eyelids, and restricted eye movement leading to diplopia.[51] Individuals may experience dull, agonizing discomfort around the eyes, exacerbated by eye movement or palpation of the orbit.
Extraocular muscle involvement can cause double vision, impairing binocular vision and ocular alignment. Nerve involvement or inflammation of the extraocular muscles may result in strabismus or ocular misalignment. Decreased visual acuity can range from slight blurring to severe vision loss due to compression or involvement of the optic nerve.
Some patients may exhibit systemic symptoms, such as arthralgias, fever, exhaustion, or weight loss, especially when autoimmune disorders or systemic inflammation are present. A frank orbital mass may be present on radiologic examination.
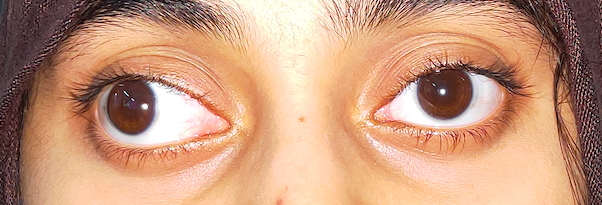
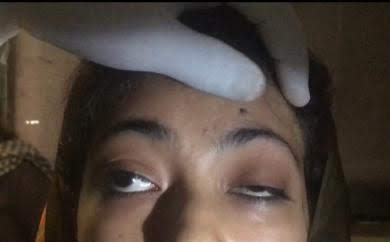
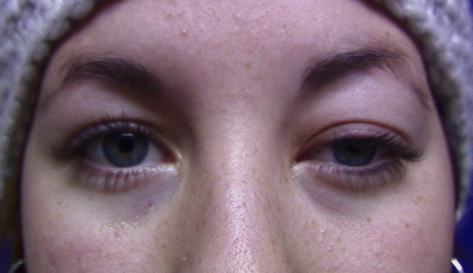
Another typical presentation is that of a patient with an acute onset of orbital pain and headache. In adults, there may be eyelid and periorbital erythema and edema, conjunctival congestion, proptosis (see Image. Proptosis), ptosis (see Image. Ptosis), diplopia, light sensitivity, decrease in eye motility, and pain on eye movement.[2][5][52] Additionally, there may be dacryoadenitis (see Image. Dacryoadenitis), myositis (see Image. Myositis), trochleitis, or diffuse soft-tissue inflammation.
In some studies, periorbital edema, ptosis, pain, and decreased extraocular muscle movements were more common in the pediatric age group.[1] Symptoms are usually unilateral in adults but bilateral in children.[5]
Dacryoadenitis accounts for 50% of all IOI’s. It typically presents with a painful, firm mass, edema in the lateral upper eyelid, and S-shaped ptosis. It may be bilateral. Some patients with idiopathic dacryoadenitis have been relabeled as having immunoglobulin G4-related ophthalmic disease (IgG4-ROD).[3][8] This recently described clinical entity reportedly affects the orbital soft tissue, optic nerve, and trigeminal nerve branches.
Orbital myositis may present as acute, subacute, or recurrent, involving one or multiple ocular muscles. The most commonly involved muscles are the medial rectus, followed by the superior, lateral, and inferior recti muscles.[1] Orbital myositis primarily affects young adults in the third to the fourth decade of life with a female predominance.[3]
Systemic disorders have been correlated with orbital pseudotumor. Immunologic diseases such as rheumatoid arthritis, Crohn disease, SLE, and scleritis have been reported to cause orbital inflammatory disease. Additionally, sinusitis has been implicated. Concurrent systemic diseases such as flu, asthma, colitis, and severe anemia have also been correlated with NSOI, as have hepatitis C, rheumatoid arthritis, and xanthogranuloma.
Physical Examination
A complete review of systemic findings should be combined with a full ocular examination to identify orbital pseudotumor correctly. Essential elements of the physical assessment include:
- Visual acuity and visual fields: Examine each eye separately for visual acuity and check for any abnormalities in the visual fields that may indicate compression or involvement of the optic nerve.
- External examination: Check for edema, erythema, or periorbital swelling. Examine the posture of the eyelids, noting any ptosis or retraction, and assess for enophthalmos or proptosis.
- Ocular motility: Examine the function of the extraocular muscles and check for any limitations in eye movements, as this may indicate inflammation or compression of the ocular motor neurons.
- Anterior segment examination: Look for indications of inflammation, such as conjunctival injection, corneal edema, anterior chamber cells, or flare, in the conjunctiva, cornea, anterior chamber, and iris.
- Fundus examination: Perform a dilated fundus examination to examine the optic nerve head, retina, and macula for signs of choroiditis, retinal vasculitis, or involvement of the optic nerve.
Evaluation
Laboratory Findings
Laboratory evaluation usually includes hematologic work-up, routine biochemistry, inflammatory markers, and a comprehensive autoimmune panel. A hematologic work-up consists of a complete blood cell count, erythrocyte sedimentation rate, C-reactive protein, electrolytes, thyroid function studies, sedimentation rate, antinuclear antibodies, antineutrophil cytoplasmic antibodies, rheumatoid factor, angiotensin-converting enzyme level, and rapid plasma regain test.[5][53] Active disease activity can be assessed by inflammatory markers such as soluble interleukin-2 receptor (sIL-2R) and interleukin-6 (IL-6).[54] While laboratory findings within normal limits yield little diagnostic value, they are necessary to exclude other causes.
Ultrasound, Computed Tomography, and Magnetic Resonance Imaging
Due to their ability to provide precise anatomical information and define the degree of orbital inflammation, imaging modalities such as CT scans and MRI are essential in assessing orbital pseudotumor. MRI, with its enhanced soft tissue contrast resolution and multiplanar imaging capabilities, is ideal for identifying related inflammatory alterations (eg, fat stranding, enhancement patterns), detecting orbital mass, and assessing extraocular muscle involvement. CT imaging helps analyze bone architecture, identify calcifications, and distinguish between inflammatory lesions and neoplastic processes.
Ultrasonography may help evaluate the globe and ocular complications such as retinal and choroidal detachments.[39] CT and MRI findings in anterior IOI include an anterior orbital mass with a molded configuration and uveoscleral thickening.[3] Enlargement of extraocular muscles on CT scans is a common finding in orbital myositis. IOI lesions enhance with contrast, and on MRI, they may display fat-suppressed T2-weighted image signals depending on the degree of tissue edema. A diffuse lacrimal involvement typically affects the orbital and palpebral lobes.[28] Optical coherence tomography (OCT) can provide information about anterior segment structures, retina, and optic nerve, which can be helpful in the diagnosis and follow-up of these patients.[55]
Enhancement is variable depending on the stage. In the acute stage, there is pronounced enhancement, while in the chronic and sclerosing type of inflammation, hypointensity on T2-W images is noted. Diffuse IOI CT and MRI findings are similar to anterior IOI. Differentiating orbital lymphoproliferative diseases may be challenging. Apical IOI may show signs of intracranial involvement, such as abnormal soft tissue extending into the middle cranial fossa, cavernous sinus, and meninges.[3] Radiologic results help classify the type of orbital tissues involved in the inflammation, including the lacrimal glands, extraocular muscles, optic nerve, orbital fat, and apex and intracranial areas.[19]
Novel diagnostic tools include imaging techniques that use artificial intelligence and deep learning.[56] Studies have shown that clinical data combined with quantitative imaging data assessed with deep learning and artificial intelligence can provide more accurate discrimination in diagnosing orbital pseudotumors.[57][58] Convolutional neural networks, which provide multilayer analysis based on deep learning models, have shown promising preliminary results as a personalized diagnostic tool in these patients.[59] Radiomics, defined as image feature extraction from a large amount of highly complex data, has shown to be useful in discriminating pseudotumors from other orbital diseases.[60] New biomarkers, like microRNA (miRNA), have also shown promise in diagnosing and evaluating treatment response.[61][62]
Treatment / Management
Current therapeutic agents available for idiopathic orbital inflammation are:
- Corticosteroids
- Nonspecific steroid-sparing agents
- Methotrexate
- Cyclosporin-A
- Mycophenolate mofetil
- Cyclophosphamide
- Rituximab
- Biologic agents
- Infliximab
- Adalimumab
- Etanercept
- Daclizumab
- Abatacept
- Tocilizumab
- Radiation therapy and surgery for nonresponders
While a modest proportion of patients experience resolution of their symptoms without treatment, orbital pseudotumor responds dramatically to steroids.[63] Systemic corticosteroids, such as prednisone or methylprednisolone, are the cornerstone of early medical treatment for ocular pseudotumor. Glucocorticoids are considered first-line treatment in these patients, given the advantages of being inexpensive, relatively effective, readily available, and providing rapid onset of clinical improvement of signs and symptoms.[64]
A "corticosteroid response" is defined as a dramatic improvement of signs and symptoms within 48 hours after the administration of systemic prednisolone.[1] Idiopathic orbital myositis typically exhibits this response. However, other types of pseudotumors are not as readily assessed by this corticosteroid trial.[28][53] Reports exist of an erroneous diagnosis of IOI based on a corticosteroid trial in lymphomas, metastatic disease, fungal infections, and sarcoidosis, among others.[37] Long-term corticosteroid usage is associated with a wide range of side effects, such as immunosuppression, diabetes, hypertension, and osteoporosis, which calls for careful monitoring and the use of steroid-sparing medications in some situations.[65]
To achieve a quick remission of inflammation, high-dose corticosteroids are frequently used, followed by a stepwise tapering regimen designed to minimize adverse effects and preserve disease control. For adults, initial steroid treatment doses are typically 1 mg/kg of prednisone. High-dose oral corticosteroids of 1.0 to 1.5 mg/kg/day are the preferred protocol for children.[66] The reported total dose ranges from 60 to 100 mg daily for 1 to 2 weeks with a 5- to 6-week taper.[49] The steroids can begin tapering as soon as the clinical response is complete.
There is no consensus on the treatment protocol for cases that do not respond to steroid treatment. The reported cure rate is only 37%, and a recurrence rate of 52% with corticosteroid therapy. Long-term use of steroids leads to systemic side effects, including insomnia, hyperglycemia, weight gain, and cataracts. Local corticosteroid injections (such as triamcinolone acetonide) for periorbital and orbital inflammation are also options.[67]
For patients who are nonresponsive to steroid treatment or suffer relapses despite steroid treatment, antimetabolites, alkylating agents, T-cell/calcineurin inhibitors, lymphocyte inhibitors, TNF-α inhibitors, and surgical debulking may be employed.[3] These treatments aim to suppress the inflammatory response and prevent further damage to orbital tissues.
Steroid-sparing medications may be considered as an additional or alternative therapy in situations of corticosteroid dependency, intolerance, or refractoriness to preserve disease management and reduce corticosteroid-related comorbidities. Immunomodulatory drugs such as methotrexate, azathioprine, mycophenolate mofetil, and cyclosporine are frequently utilized to treat orbital pseudotumor.[68] These medications facilitate corticosteroid tapering and avoid disease recurrence by lowering inflammatory cell infiltration inside the orbit and regulating aberrant immune responses.
Immunosuppressive or immunomodulatory therapy may be an option in selected cases.[69] These include cyclosporine, cyclophosphamide, methotrexate, azathioprine, mycophenolate mofetil, etc. Cyclosporin-A (CsA) suppresses the lymphocyte-mediated responses. It inhibits T-cell activation via decreased production of interleukin-1 and interleukin-2. Renal function requires monitoring while on CsA.[5] Antimetabolites like methotrexate, which inhibits dihydrofolate reductase needed for DNA and RNA synthesis, suppress rapidly dividing B-cells and T-cells.[5] Azathioprine, a purine analog that interferes with DNA synthesis, especially of immune system cells, and mycophenolate mofetil, a purine synthesis inhibitor, have had therapeutic success.[3] Alkylating agents such as cyclophosphamide and chlorambucil, which damage proliferating cells by crosslinking DNA, have been reported to be effective, though infrequently used.
For patients with orbital pseudotumor who are unresponsive to corticosteroids or who are resistant to them, biologic medicines that target specific inflammatory pathways have emerged as viable treatment choices.[70] TNF-α inhibitors such as infliximab and adalimumab have effectively lowered orbital inflammation and maintained disease remission in specific individuals. Infliximab, a chimeric monoclonal antibody that works against TNF-α, has been used for autoimmune diseases such as Crohn disease, ulcerative colitis, and rheumatoid arthritis. Rituximab, a monoclonal antibody that targets B cells positive for CD20, has also shown promising results in orbital pseudotumor, mainly when used in conjunction with autoimmune illnesses. Other therapeutic agents and immunomodulators used for refractory OIP are interferon-A, tacrolimus, rituximab (a B-lymphocyte inhibitor monoclonal antibody), daclizumab (T-lymphocyte inhibitor monoclonal antibody), and leflunomide.[5]
Radiotherapy may also be an option in individuals who are intolerant to cortisone, in refractory cases, those who are hormone-dependent, and those who are unresponsive.[71] Usually, low doses of 20 to 30 Gy at 2 Gy per fraction are utilized.[63] Treatment options include opposed lateral field 3-D conformal radiation and en-face electron intensity modulated radiation.[72]
While idiopathic orbital inflammation is generally benign, its course can be clinically fulminant with vision loss and severe oculomotor dysfunction. In cases where medical therapy is ineffective, such as refractory illness, compressive optic neuropathy, or severe vision-threatening consequences, surgical measures may be necessary.[7] Orbital decompression, achieved through either transcutaneous or endoscopic methods, aims to reduce orbital pressure and alleviate optic nerve compression, thereby preserving visual function and relieving symptoms such as proptosis or diplopia.[73]
Surgical excision or debulking of ocular lesions may also reduce tumor burden and relieve compressive symptoms in situations of mass effect or localized inflammation. Patients with optic nerve involvement may require admission for aggressive intravenous steroid therapy to prevent long-term visual impairment.[52] Thus, prompt diagnosis and treatment are imperative.
Differential Diagnosis
Diagnosing orbital pseudotumor requires careful evaluation due to the overlap of clinical features with benign and malignant orbital diseases. A systematic approach is essential to distinguish orbital pseudotumor from other disorders and determine the best course of treatment, incorporating clinical characteristics, imaging results, laboratory tests, and histological studies.
The following diseases, among others, should be considered in the differential diagnosis to avoid misdiagnosis:[28][74]
- Infections: Similar to orbital pseudotumor, orbital cellulitis is a bacterial infection of the orbit that can cause symptoms such as eyelid edema, proptosis, discomfort while moving the eyes, and reduced vision.[75] However, systemic symptoms (such as fever and malaise), an abrupt start, and signs of infection on laboratory tests (such as leukocytosis and raised inflammatory markers), as well as imaging (such as sinus opacification and orbital abscess), are usually linked to orbital cellulitis.
- Systemic diseases: Proptosis, eyelid retraction, diplopia, and abnormalities in ocular motility are among the clinical characteristics of thyroid eye illness, commonly referred to as Graves' orbitopathy, that are similar to those of orbital pseudotumor.[76] However, thyroid autoantibodies, such as thyroid-stimulating immunoglobulin, and distinctive orbital imaging findings, such as extraocular muscle enlargement with sparing of the tendons and apices, may serve as differentiators for thyroid eye disease.
- Malignancies: Malignancies like lymphoma, metastatic carcinoma, and leukemia can be clinically and radiologically associated with orbital pseudotumor, especially orbital lymphoma, a neoplastic disorder originating from lymphoid tissue inside the orbit.[77] However, distinctive characteristics such as palpable orbital masses, progressive, painless proptosis, and imaging findings such as homogeneous enhancement with contrast and involvement of orbital structures not usually affected in orbital pseudotumor can help differentiate orbital lymphoma.
- Confirmation of orbital lymphoma involves histopathological investigation showing monoclonal lymphocytic infiltrate. Additionally, when systemic cancers such as breast, lung, or prostate cancer spread to the orbits, they can cause orbital inflammation and mass effects that mimic orbital pseudotumor.[78] Patients presenting with an orbital mass, progressive, painless proptosis, or compression of the optic nerve should be clinically considered more seriously.
- Imaging examinations may reveal characteristics like numerous enhancing tumors with bone damage. The diagnosis is confirmed by biopsy when metastatic cancer is found.
- Autoimmune disorders: Systemic autoimmune pathologies such as SLE and rheumatoid arthritis can mimic signs and symptoms of orbital pseudotumor.[22] These conditions often present with systemic manifestations and specific autoantibodies, helping to differentiate them from idiopathic orbital inflammation.
- IgG4-related orbital disease: IgG4-related diseases cause fibro-inflammatory disorders that can affect the lacrimal gland, leading to swelling and enlargement.[79] This condition often presents with elevated serum IgG4 levels and characteristic histopathological features, aiding in its diagnosis.
- Sarcoidosis: Orbital sarcoidosis, a manifestation of systemic sarcoidosis, can present with orbital masses and granulomatous inflammation that resemble orbital pseudotumor. Patients with classic imaging features such as lacrimal gland enlargement, perineural enhancement, and bilateral orbital involvement, along with systemic symptoms of sarcoidosis (eg, pulmonary involvement, skin lesions), should be considered clinically suspicious. The diagnosis is confirmed by a biopsy that shows noncaseating granulomas, a hallmark of sarcoidosis.[80]
Orbital and systemic diseases that may present with similar signs and symptoms include orbital cellulitis, thyroid eye disease, sarcoid, orbital lymphoma, lymphangioma, metastatic carcinoma, leukemia, lymphoproliferative disorder, and rhabdomyosarcoma.[52] Tolosa Hunt syndrome shares similar symptoms of periorbital pain and cranial nerve palsy, and it also has an excellent response to steroids. More severe conditions, such as metastases from rhabdomyosarcoma or Ewing sarcoma, chronic recurrent multifocal osteomyelitis (CRMO), and SAPHO ( synovitis, acne, pustulosis, hyperostosis, and osteitis) have been known to cause orbital pseudotumor.
Bilateral sclerosing orbital pseudotumor should alert the physician. This subset of IOI leads to serious morbidity with a chronic, severe, progressive disease commonly manifesting with proptosis, restricted ocular motility, and pain.[3] There is emerging evidence that sclerosing IOIi is a subset of IgG4-related disease. Ocular adnexal IgG4 disease should be a consideration if there is bilateral ocular or systemic involvement.[81]
In children, the differential diagnoses include orbital cellulitis, orbital trauma with retained foreign body, ruptured dermoid cyst, lymphangioma, neuroblastoma, Langerhans cell histiocytosis, and pediatric malignancies such as rhabdomyosarcoma, leukemia, neuroblastoma and, metastatic retinoblastoma.[3] Additionally, inflammatory conditions like IOI and thyroid eye disease should also be considered in pediatric cases.
A combination of ancillary laboratory examinations, CT scans, MRI, OCT, and biopsy may be necessary to distinguish among the clinical entities that mimic orbital pseudotumor. These tools can help establish an accurate diagnosis and inform appropriate management strategies.
Prognosis
While orbital pseudotumor resolves without treatment in some patients, steroids are the mainstay of treatment. Over 75% of patients show improvement within 24 to 48 hours of steroid treatment.[2] As mentioned previously, the reported cure rate is 37%. Recurrence is approximately 52%, even with corticosteroid treatment.[67]
Factors reported to be associated with multiple recurrences include patients younger than 16 years, the presence of bilateral disease, signs of optic disc edema, sclerosing variant, poor steroid responders, and recurrence of the disease within 3 months.[36] Incomplete resolution of inflammation, corticosteroid dependency, insufficient tapering of immunosuppressive medication, and underlying autoimmune diseases are factors linked to an increased chance of recurrence. Repeated orbital pseudotumor episodes may exacerbate visual results and functional impairment by causing increasing orbital fibrosis, tissue scarring, and cumulative damage to orbital components.
The prognosis for orbital pseudotumor varies greatly based on each patient's unique features, the severity of the disease, and the patient's reaction to therapy. While many patients with proper care have positive outcomes, others may encounter complications like diplopia, corneal exposure, optic nerve compression, and disease recurrence. These cases highlight the significance of close monitoring and multidisciplinary care in achieving the best possible long-term visual and functional outcomes.
Complications
If the orbital pseudotumor is left untreated or unresponsive to treatment, vision loss, and severe oculomotor dysfunction can be permanent. The inflammation can spread to contiguous structures such as the periorbital area, the optic nerve, and the intracranial cavity. If orbital edema or inflammatory masses compress the optic nerve, optic neuropathy, and permanent vision loss may result.
Multiple cranial nerves may be affected, and sensorimotor hemiparesis may occur. Diplopia, strabismus, and poor ocular motility can be caused by the involvement of the extraocular muscles or their innervation, which can significantly negatively influence quality of life and visual function.
Severe inflammation may cause secondary angle-closure glaucoma because of choroidal effusions that cause the anterior rotation of the ciliary body.[2] Progressive proptosis may lead to exposure keratitis and ulcer formation. Patients who have proptosis and eyelid retraction due to orbital pseudotumor may be at risk for corneal exposure, corneal ulceration, and epithelial breakdown, which can lead to infection and visual impairment.
Systemic adverse effects from long-term systemic corticosteroid therapy in orbital pseudotumor care include osteoporosis, diabetes, hypertension, weight gain, and heightened susceptibility to infections. Regular monitoring for these side effects is essential to mitigate their impact and adjust treatment as needed.
Deterrence and Patient Education
Patients should be educated that a trained medical professional should evaluate symptoms of vision loss or eye pain. If orbital pseudotumor has been diagnosed, patients should also be educated about the importance of continuing treatment for orbital pseudotumor.
Patient education is essential to the therapy of orbital pseudotumor because it enables patients to take an active role in their care, follow prescribed treatment plans, identify exacerbations of their condition, and seek timely medical assistance when needed. Patients who are informed about the characteristics of orbital pseudotumor, its problems, available treatments, and prognosis make educated decisions and promote better treatment outcomes.
Pearls and Other Issues
Key facts to keep in mind about orbital inflammation include the following:
- The signs and symptoms of orbital pseudotumor may mimic those of other orbital diseases, such as autoimmune disorders, thyroid disease, or malignancy.
- A careful history and a physical examination by an internist, primary care physician, or medical specialist are particularly relevant to rule out systemic diseases that may be associated with orbital pseudotumor.
- Cooperation between ophthalmologists, rheumatologists, radiologists, and other experts is crucial to fully evaluate, diagnose, and treat orbital pseudotumor, especially in cases with refractory or unusual presentations.
- Ancillary laboratory and diagnostic testing, including CT, MRI, and biopsy, must be used judiciously for a definitive diagnosis.
- Clinicians should carefully weigh treatment options for patients who are nonresponsive to a steroid regimen.
- Since orbital pseudotumors vary significantly in their clinical presentations and prognoses, it is critical to customize treatment regimens based on each patient's unique features, the severity of their condition, and their response to therapy.
- Long-term surveillance and monitoring are required to evaluate disease activity, treatment response, and possible complications, even in patients who achieve remission, in order to identify early indicators of a return of the illness or unfavorable consequences of treatment.
Enhancing Healthcare Team Outcomes
Since orbital pseudotumor is diagnosed by excluding other diseases, the health care team should consist of an interprofessional team of pediatricians, primary care physicians, internists, rheumatologists, endocrinologists, allergologists, immunologists, radiologists, and autoimmune disease experts. A comprehensive systemic examination by qualified medical personnel and a carefully documented history are vital to the ophthalmologist's diagnosis and management.
For the therapy of orbital pseudotumors, a multidisciplinary strategy comprising several healthcare specialists, such as ophthalmologists, rheumatologists, radiologists, pathologists, and specialized nurses, is necessary. Optimizing patient outcomes requires a collaborative effort to enable thorough examination, precise diagnosis, customized treatment planning, and coordinated care delivery.
A team's communication ability is critical to smoothly transferring clinical information and coordinating care. Frequent multidisciplinary gatherings, case conferences, and electronic health record systems let team members communicate, guarantee continuity of treatment, and make joint decisions.
It is critical to prioritize patient-centered treatment when managing orbital pseudotumor. When creating treatment plans, building a cooperative therapeutic relationship, and enabling patients to actively engage in their care decisions, healthcare practitioners should prioritize their patients' preferences, values, and objectives. Systemic therapy should involve a follow-up with physicians to monitor the side effects or complications of treatment.
The patient or the patient's family should always be informed of his prognosis at each stage of the disease and be actively involved in choosing diagnostic procedures and management. Ophthalmology nurses often arrange patient follow-ups, provide education, and report to the team. Pharmacists review prescriptions for dosage and interactions. They assist the team by educating patients about the importance of compliance and discussing side effects. These are examples of how the interprofessional team approach can improve patient outcomes in orbital pseudotumor cases.
Healthcare personnel who treat patients with orbital pseudotumors must participate in ongoing education and training programs. The main focuses of training programs should be enhancing diagnostic abilities, comprehending treatment modalities, deciphering imaging studies, and remaining current with new research discoveries and evidence-based procedures.