Continuing Education Activity
Sickle cell anemia is an inherited disorder of the globin chains that causes hemolysis and chronic organ damage. Sickle cell anemia is the most common form of sickle cell disease (SCD), with a lifelong affliction of hemolytic anemia requiring blood transfusions, pain crises, and organ damage. Since the first description of the irregular sickle-shaped red blood cells (RBC) more than 100 years ago, our understanding of the disease has evolved tremendously. Recent advances in the field, more so within the last three decades, have alleviated symptoms for countless patients, especially in high-income countries. This activity reviews the pathophysiology, presentation, complications, diagnosis, and treatment of sickle cell anemia and also highlights the role of the interprofessional team in the management of these patients.
Objectives:
Describe the pathophysiology of sickle cell anemia.
Summarize the epidemiology of sickle-cell anemia.
List the management options for sickle cell anemia.
Outline the importance of cooperation among healthcare professionals to educate the patients on getting vaccinated, remaining hydrated, and timely follow-up to prevent the development of complications in those with sickle cell disease.
Introduction
Sickle cell disease (SCD) refers to a group of hemoglobinopathies that include mutations in the gene encoding the beta subunit of hemoglobin. The first description of SCA 'like' disorder was provided by Dr. Africanus Horton in his book The Disease of Tropical Climates and their Treatment (1872). However, it was not until 1910 when Dr. James B Herrick and Dr. Ernest Irons reported noticing 'sickle-shaped' red cells in a dental student (Walter Clement Noel from Grenada).[1] In 1949, independent reports from Dr. James V Neel and Col. E. A. Beet described the patterns of inheritance in patients with SCD. In the same year, Dr. Linus Pauling described the molecular nature of sickle hemoglobin (HbS) in his paper 'Sickle Cell Anemia Hemoglobin.' Ingram Vernon, in 1956, used a fingerprinting technique to describe the replacement of negatively charged glutamine with neutral valine and validated the findings of Linus Pauling.[2]
Within the umbrella of SCD, many subgroups exist, namely sickle cell anemia (SCA), hemoglobin SC disease (HbSC), and hemoglobin sickle-beta-thalassemia (beta-thalassemia positive or beta-thalassemia negative). Several other minor variants within the group of SCDs also, albeit not as common as the varieties mentioned above. Lastly, it is essential to mention the sickle cell trait (HbAS), which carries a heterozygous mutation and seldom presents clinical signs or symptoms. Sickle cell anemia is the most common form of SCD, with a lifelong affliction of hemolytic anemia requiring blood transfusions, pain crises, and organ damage.[3]
Since the first description of the irregular sickle-shaped red blood cells (RBC) more than 100 years ago, our understanding of the disease has evolved tremendously. Recent advances in the field, more so within the last three decades, have alleviated symptoms for countless patients, especially in high-income countries. In 1984, Platt et al. first reported the use of hydroxyurea in increasing the levels of HbF.[4] Since then, the treatment of sickle cell has taken to new heights by introducing several new agents (voxelotor, crizanlizumab, L-glutamine) and, most recently, gene therapy.
Etiology
Hemoglobin (Hb) is a significant protein within the red blood cell (RBC). It comprises four globin chains, two derived from alpha-globin (locus on chromosome 16) and two from beta-globin (locus on chromosome 11). There are many subtypes of Hb. The most common ones that are found in adults without hemoglobinopathies are listed here:
- HbA1- comprises two chains of the alpha-globin and two chains of the beta-globin (a2b2) - This constitutes 95% of the adult hemoglobin.
- HbA2- comprises two chains of the alpha-globin and two chains of the delta-globin (a2d2) - This constitutes less than 4% of the adult hemoglobin.
- HbF- comprises two chains of the alpha-globin and two chains of the gamma-globin (a2g2) - This Hb is more prevalent in the fetus (due to the high oxygen binding affinity that helps extract oxygen from maternal circulation).
The sickle cell mutation occurs when negatively charged glutamate is replaced by a neutral valine at the sixth position of the beta-globin chain. The mutation is transmitted via Mendelian genetics and is inherited in an autosomal codominant fashion.[5] A homozygous mutation leads to the severest form of SCD, ie, SCA- also called HBSS disease. The coinheritance of beta-naught thalassemia and sickle cell mutation leads to HBS-Beta-0 disease, which phenotypically behaves like HBSS disease.
A heterozygous inheritance leads to HbAS. Patients with HbAS are not considered within the spectrum of SCD as most of them never present with typical symptoms of SCA. They might only be detected during childbirth, blood donation, or screening procedures.
Several other compound heterozygotes exist where a single copy of the mutated beta-globin gene is coinherited with a single copy of another mutated gene. The second most common variant of SCD is the HbSC disease, where the sickle cell gene is coinherited with a single copy of the mutated hemoglobin C gene. HbC is formed when lysine replaces glutamine at the sixth position on the beta-globin chain. HbSC disease accounts for 30% of patients in the United States.
Epidemiology
The epidemiological data on SCD is scarce. It is well known that SCD and HbAS are more prevalent in sub-Saharan Africa, where the carrier of HbAS is afforded natural protection against severe Plasmodium falciparum malaria. It is estimated that ~230,000 children were born with SCA, and more than 3.5 million neonates were born with HbAS in sub-Saharan Africa in 2010. an estimated 75% of SCD-related births take place in sub-Saharan Africa. West Africa is home to the largest population of individuals with HbSC disease.[3]
The United States (US) Center for Disease Control (CDC) estimates that approximately 100,000 Americans have SCD. The CDC also estimates that 1 in 13 babies born to African-American parents have sickle cell trait, and 1 in 365 African-Americans have SCD. The estimated ratio of Hispanic Americans with SCD is 1 in 16,300. Children and adolescents make up to 40% of all SCD patients in the US. The incidence varies by state and geographical concentration of ethnicities. Besides, migration within the country and immigration from foreign countries alter the prevalence of SCD and HbAS. This is true for several countries where patients with SCD and SCA are living. Genetic studies in Brazil have also tied the origin of such patients to the slave trade originating from West Africa (Mina Coast and Angola).[6] With the improvement in technology and ease of international migration, the incidence of SCA is predicted to rise. It is estimated that the annual number of newborns with SCA will exceed 400,000 by 2050.
There is also a stark difference in mortality and morbidity in high-income and low-income countries. Adopting vaccination guidelines for children with SCD and intensive screening procedures has sharply reduced the mortality of kids with SCD between 0 to 4 years (68% drop noted from 1999 to 2002 compared to 1983 to 1986). On the other hand, in sub-Saharan Africa, 50 to 90% of children born with SCD will die before their fifth birthday. Improving the care afforded in high-income countries and targeted training of healthcare providers have improved life expectancy. However, it still lags by decades compared to matched non-SCD cohorts (54 versus 76 years - projected life expectancy, and 33 years versus 67 years- quality-adjusted life expectancy).[7]
HbSC disease accounts for 30% of all patients with SCD in the US. As with HbAS, patients with the Hb C trait (heterozygous mutation) also remain asymptomatic for most of their lives. Although considered a milder variant of SCD, HbSC disease may present with severe morbidities.[8]
Pathophysiology
Sickle cell anemia is characterized by two major components: Hemolysis and vaso-occlusive crises (VOC). The defect in the beta-globin gene makes the sickle hemoglobin (HbS) molecule susceptible to converting into rigid, elongated polymers in a deoxygenated state. The sickling process is cyclical initially, where sickle erythrocytes oscillate between the normal biconcave shape and the abnormal crescent shape (acquired under low oxygen pressure). However, there comes a time when the change becomes irreversible, and the sickle erythrocytes develop a permanent sickle shape, increasing the risk for hemolysis and VOC. All variants of SCD share the same pathophysiology leading to polymerization of the HbS component.[3]
Multiple factors inherent to sickle erythrocytes, like the low affinity of HbS for oxygen, physiologically high 2,3-diphosphoglycerate, and increased sphingokinase-1 activity, lead to deoxygenation, which promotes the polymerization of HbS. In addition to this, a high concentration of HbS, abnormal activity of the Gados channel leading to dehydration, and repeated damage to the red blood cell (RBC) membrane also increase the risk of polymerization of HbS.
Oxidative stress contributes to hemolysis by auto-oxidation of HbS, leading to erythrocyte cell membrane damage. The increased expression of xanthine dehydrogenase, xanthine oxidase, and decreased expression of NADPH oxidase increase the oxidative stress within sickle RBC. A hemolyzed cell releases free hemoglobin (scavenges nitrous oxide) and arginase 1 (competes for L-arginine) that prevent the action and formation of nitrous oxide and contribute to oxidative stress and vascular remodeling (arginase-1 converts arginine to ornithine).[3]
Besides the polymerization of the HbS and intravascular hemolysis, several other factors also contribute to vaso-occlusion. For example, the sickle RBC (expresses several adhesion molecules on the surface), free heme and Hb, reactive oxygen species, and endothelium interact with each other and with neutrophils and platelets to promote vaso-occlusion and thrombosis.
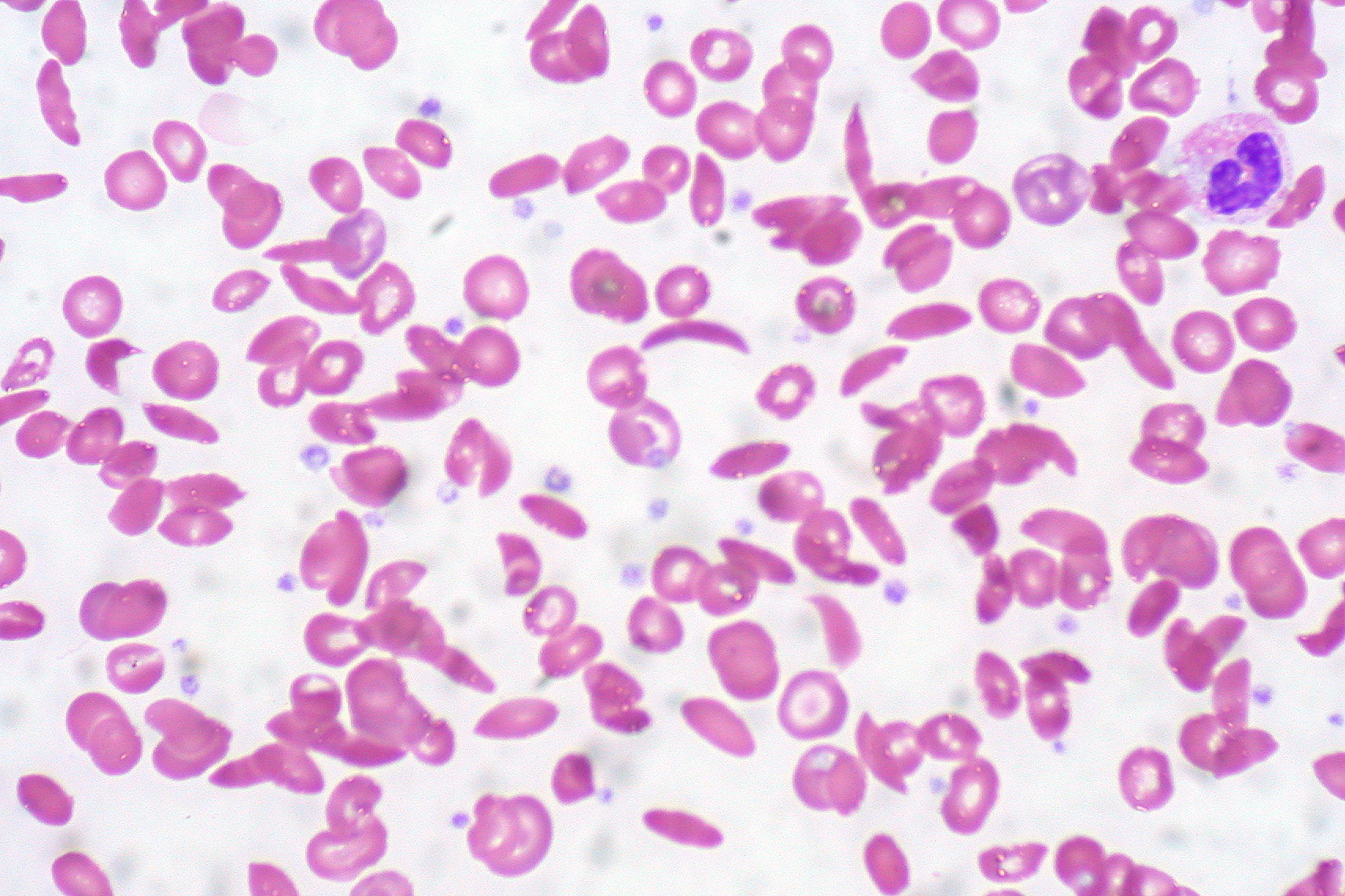
Histopathology
In patients with SCA, peripheral blood smear shows elongated RBC with tapering ends that look like a sickle (also called drepanocytes). Additional findings are present in a few patients.
- Howell-Jolly bodies- Remnants of DNA are seen in the RBC and commonly seen in patients in whom the spleen has been removed. Therefore, patients with SCA have auto-splenectomy.
- Target cells (Leptocytes)- Most commonly seen in patients with Thalassemia. They are seen frequently in sickle-thalassemia syndromes and are sometimes noted in patients with SCA.
- Polychromatic cells - these are reticulocytes that signify marrow response to hemolysis.
- Nucleated red blood cells can sometimes be visible on the peripheral smear.
None of these findings are confirmatory. Confirmation is obtained only through hemoglobin electrophoresis, high-performance liquid chromatography, or isoelectric focusing. DNA-based techniques are not used routinely. Instead, they are used in patients with uncertain diagnoses. Pre-natal fetal testing involves using fetal DNA obtained through amniocentesis. Techniques to capture the fetal DNA in maternal blood remain investigational.
History and Physical
Most patients with HbSS phenotype do not present with classical 'sickle cell crises' soon after birth. HbF is still present in the blood, helping maintain adequate tissue oxygenation, and it takes around 6-9 months to wean off completely. Not all SCAs have the same phenotype, and multiple phenotypes exist that can either co-exist or present as a spectrum of the disease.[3]
- Vaso-occlusive subphenotype - Distinguished by higher hematocrit (Hct) compared to other SCA. A higher Hct leads to higher viscosity that promotes frequent vaso-occlusive crises and acute chest syndrome.
- Hemolysis and vascular subphenotype: Lower Hct compared to other SCA, higher lactate dehydrogenase (LDH), serum bilirubin- implies a higher degree of hemolysis and severe anemia
- Higher risk of gallstones, pulmonary hypertension, ischemic stroke, priapism, and nephropathy
- Severe anemia increases cardiac workload and blood flow through organs, making them susceptible to damage.
- Higher free heme and Hb in blood vessels cause oxidative damage
- High Hb F subtype- A 10 to 15% level of HbF alleviates the symptoms of SCA. However, the distribution of HbF is not consistent throughout the body.
- Pain-sensitive subphenotype- Altered neurophysiology amongst various individuals makes them susceptible to pain. Some individuals are more susceptible to pain compared to others with SCA.
The patients with SCA present with acute or chronic complications associated with the disease. The most common acute complication of SCA is vaso-occlusive crisis (VOC). The treatment section below discusses the management of acute and chronic issues.
Important points to be noted in the history of patients with SCA
- All patients with SCA will experience VOC during their lives. The earliest presentation is dactylitis in kids as young as six months of age.
- Any body organ can develop VOC (head, eyes, etc.), although extremities and the chest are most commonly involved. If a VOC pain sounds atypical, obtain a history to rule out other causes.
- A thorough history of pain management must determine the best analgesics for the patient.
- When was the last pain crisis, and how many times in the previous year have they been admitted to the hospital with pain crises?
- If they take analgesics daily, it is prudent to know the type and quantity of the analgesic (opioid or non-opioid), the last use of analgesics, and whether they take the analgesics before coming to the ER/office.
- History of taking disease-modifying drugs (hydroxyurea, voxelotor, crizanlizumab, etc.)
- A history of substance abuse, psychiatric disorders, and use of psychotropic medications must be obtained.
- History of receiving blood transfusions and exchange transfusions- helps assess the risk of iron overload, presence of alloantibodies (multiple transfusions in the past can lead to the development of alloantibodies, which will help assess the risk of transfusion reactions), and previous transfusion reactions.
- History of any other diseases that may or may not be associated with SCA - previous history of stroke, thrombosis, priapism, etc.
- It is also advised to get in touch with the primary hematologist taking care of the patient- it is valuable to have their input in understanding the patient's normal physiology.
- History of previous surgeries.
- History of life-threatening crises in the past- if present, should alert the clinician to ensure that a similar event is not occurring again. For example, fat embolism may occur more frequently in patients with SCA.
The physical exam should focus on the general system exam to determine the need for oxygen requirements, pain management, and blood/exchange transfusion. However, a focused exam is necessary to rule out any organ-specific problem. For example, a rapidly enlarging liver or spleen should alert the physician about sequestration crises.
Evaluation
Patients with SCA are usually diagnosed in childhood. Intensive newborn screening programs in developed countries can identify patients in the neonatal stage. In the US, universal screening for SCA was implemented in all states by 2007. High-performance liquid chromatography and isoelectric focusing are the methods used in the US. In Europe, most countries deploy targeted screening in high-risk areas (where SCA is more common) and not a universal screen. In sub-Saharan Africa, no country has adopted a screening program. In India, the solubility test is used as the first step- if positive, then high-performance liquid chromatography is used to confirm at the reference center.[3]
Acute Complications in Patients with SCA
Acute Chest Syndrome (ACS): ACS is the most common complication of SCA. It is also the most common cause of death and the second most common cause of hospital admission. A patient can either present with ACS or may develop it during hospitalization for any other reason. Hence, it is prudent to monitor all patients with SCA admitted to the hospital for ACS. It is important to recognize ACS early and act upon it to prevent respiratory failure.
- The risk factors for ACS include a previous history of ACS, asthma, or recent events like recent surgical procedures, pulmonary embolism, fluid overload, infection, etc.
- The clinical features include sudden onset of cough and shortness of breath. Fever may or may not be a part of the spectrum of presentation. If present, then it usually points towards infection.
- Laboratory evaluation includes a complete blood count with differential chemistries, including liver and kidney evaluation, blood cultures, and sputum cultures.
- Chest X-ray shows a new pulmonary infiltrate- this is a quintessential feature of defining ACS. CT and perfusion mismatch scans are only used if there is a strong clinical suspicion of pulmonary or fat embolism. Therefore, they are not usually helpful in acute settings.
Sequestration Crises: This can either be hepatic or splenic sequestration.
- Splenic sequestration is a significant cause of acute anemia.
- Patients experience rapid spleen enlargement associated with pain in the left upper quadrant. In children with SCA, it is common in children between 1 to 4 years of age, as the spleen is still intact.
- Patients with non-SCA variants (HbSC, HbS-beta+ thalassemia) are not prone to 'auto-splenectomy' commonly seen in patients with SCA. Hence, they can develop splenic sequestration later in life. Such patients may have baseline splenomegaly, causing hypersplenism. Parents and patients must receive counseling regarding the signs and symptoms of an enlarging spleen.
- Younger patients present with acute anemia and hypovolemic shock due to smaller circulating volumes, whereas adults may present with a more insidious onset.
- Pain occurs due to stretching of the splenic capsule and new infarcts.
- Blood count shows a drop in Hb by more than 2gm/dL, increased reticulocyte count, and nucleated red blood cells.
- Hepatic sequestration: Hepatic sequestration can occur across all phenotypes of SCA. Like the spleen, patients may have a baseline enlargement of the liver. Hepatic sequestration is also defined as rapid enlargement of the liver with stretching of the capsule. The hemoglobin shows a drop of more than 2gm/dL. Liver enzymes may not get elevated.
Acute Stroke: Stroke is the most devastating complication of SCA. Since the advent of transcranial Doppler (TCD) and the institution of primary prevention programs, the incidence of stroke has gone down in patients with SCA. In the absence of primary prevention, ~10% of children suffer from overt stroke, and approximately 20 to 35% have silent cerebral infarcts. TCD is not useful for adults.
- Severe headache, altered mental status, slurred speech, seizures, and paralysis- are signs of stroke.
- Urgent neurological consultation and CT scan followed by MRI/MRA must be done.
Aplastic crises: It is usually precipitated by parvovirus B-19 and is defined as a rapid drop in Hb at least 3 to 6 gm/dL below the baseline. Patients present with severe fatigue, anemia, shortness of breath, and even syncope. Blood counts show severely low hemoglobin with near-absent reticulocytes. Bone marrow biopsy shows arrest in the pro-normoblast stage in patients with acute parvovirus infections.[9]
Acute intrahepatic cholestasis (AIC): Presents with sudden onset right upper quadrant pain. Physical exam shows worsening jaundice, enlarging and tender liver, and clay-colored stools. Labs show very high bilirubin levels, elevated alkaline phosphatase, and coagulopathy. The hemolysis parameters may be normal. AIC is a medical emergency.
Infections in patients with SCA can be a harbinger of infection with Streptococcus pneumoniae infection or osteomyelitis.
- The use of prophylactic antibiotics and pneumococcal vaccinations has reduced their incidence. However, loss of splenic function in SCA patients puts them at risk of invasive bacterial species.
- Osteomyelitis can be unifocal or multifocal- Staphylococcus aureus, Salmonella, and other enteric organisms can cause osteomyelitis in SCA patients.
Priapism is defined as a sustained, unwanted, painful erection lasting more than 4 hours. It is a common condition among patients with SCA, affecting 35% of all men/boys.
Acute Ocular Complications
- Hyphema- Accumulation of blood in the anterior chamber of the eye that occurs due to blunt trauma.
- The complication presents similarly in patients with SCA and sickle cell trait.
- The low oxygen pressure and acidotic nature of the aqueous humor promote sickling of the RBC, which leads to blockage of the trabecular network and an acute rise in intraocular pressure (IOP).
- High IOP is poorly handled in patients with SCA - which can lead to CRAO and secondary hemorrhages.
- Central retinal artery occlusion (CRAO)- Results from thrombus formation in the retinal artery leading to infarction of the retina, macular ischemia, or macular infarction. CRAO can occur spontaneously or secondary to increased IOP (from hyphema), Moyamoya syndrome, or ACS in patients with SCA.
- Orbital infarction: A rare complication that results from infarction of the surrounding orbital bone, which leads to an inflammatory response and compression of surrounding structures.
- Patients present with proptosis, local pain, and edema of the lid or orbit.
- The exam shows reduced extraocular motility and decreased visual acuity.
- CT scan helps in distinguishing this from orbital cellulitis/ infection.
- Orbital Compression Syndrome (OCS) - also called orbital apex syndrome, is characterized by ophthalmoplegia and vision loss secondary to events occurring at the orbital apex. Cranial nerves II, III, IV, VI, and the first division of CN V can be involved. MRI of the orbits is the best modality for diagnosis.
Chronic Complications in Patients with SCA
Iron Overload: Iron (Fe) overload is a common problem in SCA patients due to repeated transfusions and chronic hemolysis. Each unit of packed RBC contains 200 to 250 mg of iron. Excessive iron mainly affects the heart, lungs, and endocrine glands.[10] Hepatic cirrhosis from excessive iron is a major cause of death in patients with SCA. Clinical trials in patients with thalassemia have shown that systemic iron load correlates directly with survival and cardiac incidents.[11]
Avascular Necrosis (AVN) of Joints: AVN of the femoral head is a common cause of chronic pain and disability in SCA patients. Although the hip joint is the most common joint to be involved, other joints can also be affected. AVN occurs at the distal portion of the bone, where collateral circulation is poor. The capillaries get occluded by sickle RBCs, leading to hypoxia and bone death. Risk factors for AVN of the femoral head include age, frequency of painful episodes, hemoglobin level, and alpha-gene deletion. In patients with HbSS, the overall prevalence is 50 percent by age 33. HbSS-alpha thalassemia and HbSS-Beta-0 thalassemia are at higher risk of developing AVN early in life.
Leg Ulcers: More common in SCA compared to other SCD genotypes. Approximately 2.5% of patients with SCA above ten years of age have leg ulcers. Leg ulcers are more common in men and older people and less common in people with high total hemoglobin, alpha-gene deletion, and high levels of HbF. Trauma, infections, and severe anemia also increase the risk of leg ulcers. The ulcers occur more commonly on the medial and lateral surfaces of the ankles. They vary in size and depth, and chronic ulcers may lead to osteomyelitis, especially if they are deep enough to expose the bone.
Pulmonary Artery Hypertension (PAH): Affects 6 to 11% of patients with SCA. PAH in SCA is classified under World Health Organization (WHO) group V. However, chronic hemolysis leads to pulmonary vascular changes classified under WHO group 1 in up to 10% of all SCA patients. PAH in SCA can also occur due to left heart dysfunction (Group II), chronic lung disease from SCA (Group III), chronic thromboembolism (Group IV), or extrathoracic causes (Group V).
The patient may complain of dyspnea on exertion, swelling in the legs, or present with symptoms of underlying disease (like a history of thrombosis, heart failure, etc.). An echocardiogram helps in estimating the tricuspid regurgitant jet velocity (TRV). Elevated TRV is associated with increased mortality in adults. However, TRV can be transiently elevated during acute chest syndrome. Serum NT-pro-BNP is directly correlated with mortality as well. The final diagnosis is made with a right heart catheterization.
Renal complications: Chronic kidney disease (CKD) occurs in approximately 30% of adult patients with SCA. The acidotic, osmotic, and hypoxic environment of the kidney increases the risk of polymerization of HbS, leading to the sickling of RBC. SCA patients secrete excessive creatinine in their proximal tubules. Hence, it becomes challenging to identify early signs of kidney disease, as creatinine takes a longer time to rise. Microalbuminuria (30-300mg albumin in 24-hour urine collection) is often the first manifestation of CKD. Spot urine-creatinine ratio is not validated in SCA patients due to hypersecretion of creatinine.
- Hypoesthenuria- Inability to concentrate urine due to loss of deep juxtamedullary nephrons. It is the most common complication in SCA patients. It leads to frequent urination and increases the risk of dehydration. It also increases the risk of enuresis in children.
- Renal papillary necrosis occurs due to obstruction of the vessels supplying the vasa recta, resulting in medullary infarction. It presents with hematuria. It is more common in patients with HbSC disease.
- Asymptomatic Proteinuria: It is present in 15 to 50% of patients. It develops early in life due to hyperfiltration and loss of selectivity for albumin.
Ophthalmologic Complications: Chronic eye complications are more common in patients with HbSC and HbSS disease. They are found in up to 50% of patients.
- Proliferative Sickle Retinopathy occurs due to vaso-occlusion of vitreal arterioles, leading to ischemia which leads to neovascularization. Neovascular tissue is predisposed to hemorrhage and vitreal traction forces, resulting in vitreal hemorrhage (the most severe complication of proliferative sickle retinopathy).
Treatment / Management
Patients with SCA present with acute and chronic complications.
Management of Acute Complications
Pain management is a critical part of SCA. It is challenging for clinicians to accurately assess patients' needs, especially if they meet them for the first time. Patients with SCA often suffer from the stigma of requiring high doses of opioids for pain control, which leads to them being labeled as 'opioid abusers,' 'manipulators,' or even' drug seekers.' [12]
- Analgesic administration starts simultaneously with evaluating the cause, ideally within 30 minutes of triage and 60 minutes of registration.
- Develop individualized pain management plans - this should be made available to the emergency room and should be implemented each time the patient presents with VOC and pain.
- NSAIDs are used in patients with mild to moderate pain who report prior episodes of relief with NSAIDs
- Opioids:
- Any patient presenting with severe pain- preferably used parenteral opioids. An intravenous route is preferred; however, if access is difficult, use the subcutaneous route.
- The dose of parenteral opioids is calculated based on the total dose of short-acting oral opioids taken at home.
- Pain should be reassessed every 15 to 30 minutes, and readminister opioids if needed. The escalation of opioids is done in 25% increments.
- Patient-controlled analgesia (PCA) is preferred. If an "on-demand" setting is used in PCA, then continue long-acting analgesia.
- When pain control is achieved, "wean off" parenteral opioids before converting to oral medications.
- Calculate the inpatient analgesic requirement at discharge and adjust home doses of short and long-acting opioids accordingly.
- Meperidine is not used in managing VOC-related pain unless this is the only medication that controls the pain.
- Antihistamines only help in controlling opioid-related itching. When required, use oral formulations only—readminister every 4 to 6 hours as needed.
- Supportive measures should be instituted along with pain management
- Incentive spirometry
- Intravenous hydration
- Supplemental oxygen is needed only if saturation drops below 95% on the room air.
Management of Chronic Pain
Chronic pain management in SCA patients focuses on the safe and adequate use of pain medications, particularly opioids. A comprehensive assessment of the patient's ailment, the kind and doses of pain medicine required to control pain, and the functional outcomes of using these medications are made at each encounter. The process involves collaboration with multiple specialties, like psychiatry, social work, etc., to administer the right pain medicine in the proper doses.
The strategy adopted in the clinic to prescribe pain medicine involves:
- One person must be assigned to prescribe long-term opioids. They should document all encounters extensively involving the physical exam, lab work, etc.
- Assess each patient for non-SCA-related pain and treat/refer to the appropriate specialty for managing this pain.
- Limit prescribing pain medicines without meeting the patient- every patient must be physically assessed every 2 to 3 months or sooner.
- Develop an individualized pain management plan for each patient, reassess this plan annually, and modify it accordingly.
- Encourage patients to explore alternative methods of controlling pain, like direct massage, self-hypnosis, and music therapy.
Acute Chest Syndrome (ACS): It is an emergency regardless of the sickle cell disease phenotype. It can lead to respiratory failure and death if not managed as an emergency.
- All patients must be hospitalized-
- Upon admission, start treatment with antibiotics, including coverage for atypical bacteria.
- Supplemental oxygen is provided to those with oxygen saturation of less than 95% at room air.
- "Early" administration of simple blood transfusion is recommended for hypoxic patients. However, exchange transfusion is recommended at the earliest opportunity.
- Close monitoring for worsening respiratory status, increasing oxygen requirement, worsening anemia, and bronchospasm (use of beta-adrenergic dilators is encouraged in asthmatics) must be done. Intensive care units must be on standby to receive such patients who experience worsening respiratory status.
- Closely monitor predictors of severity- increasing respiratory rate, worsening hypoxia, decreasing hemoglobin or platelet count, multilobar involvement on chest X-ray, and developing neurological complications.
- Incentive spirometry and hydration (intravenous or oral) must always be encouraged.
- ACS is a strong indicator for initiating disease-modifying therapy (hydroxyurea, etc.) or starting the patient on a chronic blood transfusion program.
Sequestration Crises
- Intravenous fluids for hydration, pain control, and simple/exchange blood transfusion are central to managing sequestration crises.
- Never correct anemia completely- when the crises resolve, and the organs shrink, the sequestered blood re-enters the circulation, leading to increased hematocrit and viscosity, increasing the risk of thrombotic and ischemic events.
- Splenectomy is recommended for patients with life-threatening episode splenic sequestration crises or with recurrent splenic sequestration. It is also offered to those who have baseline hypersplenism.
- Instruct patients and parents in monitoring the size of the liver and spleen regularly.
Acute Stroke: Urgent neurology and transfusion medicine consultation are needed to provide optimal care and prevent long-term damage.
- Simple or exchange blood transfusion emergently.
- Start a program of chronic exchanges or blood transfusion.
- Where blood transfusion cannot be used (iron overload, excessive alloantibodies) or is unavailable, start on long-term disease-modifying therapy. SWiTCH trial demonstrated that chronic transfusions are a better way to manage patients with stroke.
Aplastic Crises: Parvovirus infections cause a transient drop in hemoglobin. Humoral immunity develops within 7 to 10 days that stays for life. The patient is extremely susceptible to developing ACS or stroke during the acute period. Initiate exchange/simple transfusion to bring Hb to a safe level, not necessarily to normal/baseline level.
Infections presenting with fever: Oral empiric antibiotics are given promptly while evaluating the reason for the fever. For ill-appearing patients, admit them and administer intravenous antibiotics.
Priapism: Early recognition is the key to management. Delayed management can lead to impotence. Urologists need to be involved early on in the care of such patients.
- Conservative measures include using analgesics, hydration, and sedation - which usually leads to detumescence and retains potency. Most experts would call for upfront urologic management rather than losing time trying conservative measures.[13]
- Urologists can perform penile aspiration or irrigation of corpora cavernosa with alpha-adrenergic drugs.
- Blood transfusion/ exchange transfusion is not useful - few authors have reported neurological complications with the use of blood transfusion (ASPEN syndrome). Hence it is best to avoid blood transfusion.
Acute ocular Complications: All ocular complications must be managed in consultation with ophthalmologists and hematologists to prevent vision loss.
- Hyphema- Anterior chamber paracentesis or surgical intervention to manage the thrombus must be done promptly.
- Reducing intraocular pressure helps prevent CRAO and other compression issues.
- Infections are managed with prompt administration of antibiotics.
- Corticosteroids are used to relieve excessive pressure in patients with OCS.
Chronic Complications
Avascular Necrosis: About 40 to 80% of cases of hip joint AVN are bilateral; therefore, both joints should be investigated simultaneously. Pain management and physical therapy are to be initiated as early as possible. Advanced cases may require hip arthroplasty.
Leg Ulcer: Conservative measures involve wound care, wet-to-dry dressings, and pain control. Hydroxyurea is avoided in patients with open leg ulcers, as it may prevent healing. Frequent evaluation for the stage of healing or lack of infection, osteomyelitis must be done. Local and systemic antibiotics are used for infected ulcers.
Pulmonary Hypertension: Patients with higher TRV are referred to pulmonologists for management. Small studies have shown increased mortality with sildenafil.
Renal Complications: Refer SCA patients with micro- or microalbuminuria to nephrologists for detailed workup and consideration of angiotensin-converting enzyme inhibitor (ACE-inhibitor). Follow patients closely who have modest elevation in creatinine (>0.7 mg/dL in children, >1.0 mg/dL in adults), and refer to a nephrologist at the earliest sign of worsening creatinine.
Ophthalmologic Complications: Refer SCA patients regularly for ophthalmologic evaluation, especially if they complain of slow vision changes. Direct and indirect ophthalmoscopy, slit-lamp biomicroscopy, and fluorescein angiography are used to evaluate SCA patients. Laser photocoagulation therapy is used to manage proliferative sickle retinopathy. A vitrectomy or retinal repair may be needed in the rare event of vitreal hemorrhage or retinal detachment.
Iron Overload
Unlike hemochromatosis, phlebotomy is not an option in patients with SCA. Preventing iron overload with good transfusion practices is the best way to deal with iron overload. Patients with SCA need not follow the rule of having hemoglobin close to 7gm/dL. Packed RBC transfusion should be restricted to the management of symptoms. Choosing exchange transfusion over simple transfusion also helps to reduce/prevent iron overload.
Indications to start iron chelation therapy
- A liver iron concentration (LIC) greater than 3 mg iron (Fe)/gm dry weight
- Cardiac T2* < 20 milliseconds
- Serum ferritin greater than 1000 on two different occasions 15 days apart
- Populations that should be considered for iron overload therapy
- Age greater than two years
- Expected survival beyond one year
- Number of transfusions of packed RBC in 1 year- > 10 in pediatric patients OR > 20 in adults.
Goals of therapy
- Serum ferritin < 1000 mcg/L,
- LIC <7mg Fe/gm dry weight
- Cardiac T2* > 20 milliseconds
When do patients need modification of treatment?
- Treatment needs to be intensified if LIC > 15 mg Fe/gm dry weight and deescalated when LIC < 3 mg Fe/gm dry weight.
- Treatment needs to be intensified if serum ferritin > 2500 IU/L and deescalated when serum ferritin < 300 IU/L
- Treatment needs to be intensified when cardiac MRI shows T2* < 15 milliseconds or when cardiac symptoms occur (like heart failure, arrhythmias)
Iron Chelators
- Deferasirox:
- Disperse tab formulation: Initial dose: 10mg/kg/day. Maximum dose: 20mg/kg/day
- Tablet or granule formulation: Initial dose: 7mg/kg/day. Maximum dose: 14mg/kg/day
- It does not interfere with the pharmacodynamics of hydroxyurea; hence it can be used simultaneously.
- Adverse effects- gastrointestinal intolerance, dose-dependent rise in serum creatinine, liver dysfunction.
- Deferoxamine injection:
- Daily subcutaneous infusions via portable infusion pump given over 8 to 24 hours; 1 to 2 gm/day
- It can be given as a daily IV infusion also. 40 to 50 mg/kg/day (max dose 60 mg/kg/day) over 8 to 12 hours (max rate 15 mg/kg/hour)
- IM route is acceptable for children but not preferred for adults. 0.5 to 1mg/day
- Adverse effects- Injection site reactions, cardiovascular shock (if administered too fast), blood dyscrasias, growth retardation.
- Deferiprone (Approved for thalassemia only); oral drug NOT approved for SCA by US FDA.
- Adverse effects - agranulocytosis, hepatotoxicity, gastrointestinal symptoms, and arthralgia.
Blood transfusion: Blood transfusions form an integral part of the management of SCA. The goal of transfusion is to increase the oxygen-carrying capacity of blood and reduce the HbS component. A blood transfusion (simple or exchange) is given to keep the HbS level below 30% (STOP 1 and 2 trials).[14] In patients receiving regular exchange transfusions (history of stroke, intolerance, or contraindication to hydroxyurea), a more practical target for HbS is 25% to prevent a rise of HbS beyond 30%.
What types of blood transfusion are used in SCA?
- Simple transfusion: Transfusion of matched packed red blood cells (PRBC)
- Exchange transfusion: Transfusion of PRBC while removing blood from the patient at the same time.
Who should receive blood transfusions?
- Pregnant females have high maternal-fetal morbidity if Hb is below 7 gm/dL.
- Hb < 7gm/dL or drop of >2 gm/dL from baseline- consider simple or exchange transfusion.
- Twin pregnancy- consider prophylactic exchange transfusion
- Preoperative transfusion for medium-risk surgery (cholecystectomy, joint replacement)- transfuse to maintain Hb above 10gm/dL
- Hb less than 9 gm/dL- Simple transfusion
- Hb more than 9gm/dL- Partial exchange transfusion
What kind of transfusion practice should be followed?
- Exchange transfusion
- Severe ACS - oxygen saturation less than 90% even when started on supplemental oxygen.
- Multiorgan Failure
- Acute ischemic stroke
- Simple transfusion
- Splenic sequestration - never corrects the anemia completely.
- Acute anemia
Complications from Chronic Transfusions
- Alloimmunization- increases the risk of transfusion reactions, especially delayed hemolytic transfusion reactions.
- Iron overload
- Transmission of blood-borne diseases like hepatitis B, C, and HIV; extremely low risk due to intensive screening of donors and blood products.
Differential Diagnosis
In general, globin gene mutations affecting hemoglobin are common and affect 7% of the entire world population.[15] Over 1000 variations of hemoglobin exist. However, only a handful of variations are significant clinically.
Common Variants of SCA or HbSS Disease
- Hemoglobin S-beta-0 thalassemia (Clinically behaves exactly like HbSS disease)
- Hemoglobin SC (a milder variant of SCD) - can have a phenotypic presentation of sickle cell anemia.
- Hemoglobin S-beta+ thalassemia (a milder variant of SCD)
Several other hemoglobin variants are present that can mimic SCA if they are inherited along with HbS.
- Hemoglobin Jamaica-Plain (beta-68 [E12] Leu -> Phe)
- Hemoglobin Quebec-Chori (beta-87 [F3] Thr > Ile)
- Hemoglobin D-Punjab (beta-globin, codon 121, glutamine to glutamic acid)
- Hemoglobin O-Arab
- Hemoglobin E
Other conditions that can present with hemolysis, where SCA can be ruled out with history, examination, hemoglobin electrophoresis, and study of the peripheral smear
- Antibody-mediated autoimmune hemolytic anemia (both warm and cold antibodies)
- Other hemoglobinopathies- alpha or beta-thalassemia
- Paroxysmal nocturnal hemoglobinuria
- RBC-membrane defects (hereditary spherocytosis, hereditary elliptocytosis)
- Enzyme defects (pyruvate kinase deficiency, glucose-6-phosphate deficiency)
- Drug-induced hemolysis
- Transfusion-related hemolysis (acute or delayed hemolytic reaction)
- Microangiopathic hemolytic anemia (atypical or typical hemolytic uremic syndrome, thrombotic thrombocytopenic purpura)
- Infectious causes (malaria, babesiosis, Rickettsia, Clostridia, Bartonella)
- Vasculitis-induced hemolysis
Medical Oncology
The goal of disease-modifying therapy in sickle cell anemia is to reduce the frequency of vaso-occlusive crises (VOC) and pain crises and prevent organ damage. These medications usually do not have a role "during" acute crises. Hydroxycarbamide, or hydroxyurea, was the first drug approved by the FDA for use in patients with SCA. However, the USFDA approved hydroxyurea for pediatric patients two years and above only in 2017 (based on the ESCORT HU trial).
Disease-Modifying Drugs/Therapy
The goal of disease-modifying therapy in patients with SCA is to alter the kinetics of sickle erythrocytes. Hydroxyurea does this by increasing the concentration of fetal hemoglobin (HbF).
Hydroxyurea: This is a ribonucleotide reductase inhibitor that increases the concentration of HbF in patients with SCD. It not only increases the intracellular concentration of HbF but also increases the number of erythrocytes containing HbF. In addition to this, hydroxyurea also reduces the number of circulating reticulocytes and leukocytes, raises the volume of an RBC (high MCV is noted in patients receiving hydroxyurea), reduces the deformability of RBC, improves the flow of blood through capillaries, and alters the expression of adhesion molecules hence preventing vaso-occlusive crises. The initial trials with hydroxyurea (Phase-III Multicenter Study of Hydroxyurea in Sickle Cell Anemia (MSH)) demonstrated a clear benefit over placebo in reducing the incidence of pain crises and the cost of care. Long term, the MSH study also showed a mortality benefit. In the pediatric age group, two seminal trials (HUG-KIDS-Phase I/II and BABY HUG-phase III) demonstrated good tolerability and led to the drug's approval.[16][17]
- Who should receive hydroxyurea?
- Having three or more sickle cell-associated moderate to severe pain crises within a 12-month period; treat with hydroxyurea.
- Those with sickle cell-associated pain that interferes with daily activities of living and quality of life
- History of severe and/or recurrent ACS
- Severe symptomatic chronic anemia that interferes with daily activities or quality of life
- Infants 9 months of age and older, children, and adolescents with SCA offer hydroxyurea regardless of clinical severity to reduce SCA-related complications (e.g., pain, dactylitis, ACS, anemia)
- For those with chronic kidney disease, taking erythropoietin and hydroxyurea can be added to improve anemia.
- DO NOT give hydroxyurea to pregnant women and lactating mothers who choose to breastfeed their babies.
- Clinical use of hydroxyurea:
- Dosing for adults: Start with 15 mg/kg/day. Round up to the closest 500 mg. For patients with CKD- start at 5 to 10 mg/kg/day.
- Dosing for infants and children: start at 20 mg/kg/day
- Increments are to be done every eight weeks in aliquots of 5mg/kg to a maximum of 35 mg/kg/day.
- Target absolute neutrophil count (ANC) above 2000/microL and platelet count above 80,000/microL. In younger patients, an ANC of 1250/microL is allowed if baseline counts are low.
- Monitor blood counts every four weeks when increasing the dose of hydroxyurea.
- Clinical response takes 3 to 6 months to develop. Hence, a minimal trial of 6 months of daily continued use of hydroxyurea is conducted before considering alternative therapies.
- Daily adherence is a must. It must be emphasized to the patient.
- If a positive response is seen, then hydroxyurea must be continued indefinitely.
- Adverse events:
- Myelotoxicity is the most common and most substantiated adverse effect of hydroxyurea. The rest of the adverse effects reported in the literature, especially carcinogenesis and leukemia, have never been demonstrated in large studies.
- Avoid the use of hydroxyurea in patients with leg ulcers.
Voxelotor: Voxelotor acts by inhibiting the polymerization of HbS and increasing the affinity for oxygen. It is dosed at 1500 mg by mouth daily and is approved for SCA treatment in patients 12 years of age and older. Voxelotor can be given with or without hydroxyurea. USFDA approved it in 2019 based on the results of the phase 3 HOPE trial (Hemoglobin Oxygen Affinity Modulation to Inhibit HbS Polymerization) evaluating voxelotor (1500 mg versus 900 mg versus placebo in 1:1:1 design).[18][19]
The most common adverse reactions are headache, diarrhea, abdominal pain, nausea, fatigue, rash, and pyrexia. Voxelotor interferes with high-performance liquid chromatography (HPLC). Hence, the hemoglobin quantification is not accurate when the patient is on voxelotor. HPLC should be done when the patient is off therapy. Also, the use of voxelotor may increase the Hb, but there is no evidence to suggest discontinuation of exchange transfusion in patients receiving this for stroke prophylaxis.
Crizanlizumab: A humanized immunoglobulin G2-Kappa monoclonal antibody inhibits P-selectin, thereby blocking its interaction with P-selecting glycoprotein-1. This leads to reduced interaction between activated endothelium, platelets, leukocytes, and sickled RBCs, leading to reduced VOC.[20] The phase II SUSTAIN trial demonstrated a clinical benefit of Crizanlizumab by demonstrating a reduction in pain crises, VOC, emergency room visits, and increased median time to first crises. Although the hospitalization rate was numerically lower in the intervention group, the difference was not statistically significant compared to the placebo group.[21]
It is approved for the treatment of SCA in patients 16 years of age and older. It is dosed as a 5 mg/kg intravenous infusion administered over 30 minutes at weeks 0 and 2 and then every four weeks. The most common adverse reactions are nausea, arthralgia, back pain, and pyrexia. Infusion-related reactions can occur. Crizanlizumab can interfere with platelet counts; send the blood immediately before administration or in citrated tubes.
L-Glutamine: Glutamine is the most abundant amino acid in the body. It is not an essential amino acid under normal circumstances, but in patients with SCA, a high hemolysis rate increases the demand for glutamine. L-glutamine is available in a medical formulation. The exact mechanism of action of L-glutamine remains anecdotal. It is believed to work by scavenging for reactive oxygen species and acting as a substrate for the regeneration of nitrous oxide, NAD, and NADH.[22] The USFDA approved L-glutamine in 2017 after positive results from the phase III trial. The authors demonstrated a statistically lower number of pain crises, fewer hospitalizations, fewer cumulative days in the hospital, prolonged time to first and second pain crises, and a reduced number of ACS.[23] Adverse events include constipation, nausea, headache, abdominal pain, cough, extremity pain, back pain, and chest pain. There is an additional concern that L-glutamine may increase mortality and the rate of multiorgan failure. However, these are yet exploratory.
Hematopoietic Stem Cell Transplant
Allogeneic hematopoietic stem cell transplant (HSCT) is a potentially curative option in SCA patients where cure rates approach approximately 90%. Improving the quality of life and reducing the cost of managing long-term complications trumps the cost of performing allogeneic HSC. Pre-school age is considered the best time to perform HSCT, with increased mortality recorded in older patients. A myeloablative or a non-myeloablative regimen can be used; however, myeloablative regimens are not recommended for adults. A matched sibling donor is preferred for performing allogeneic HSCT. Due to the lack of matched sibling donors, other approaches like a matched unrelated donor, umbilical cord blood transplant, and haploidentical transplant are also being explored.[24][25]
Potential barriers to performing allogeneic HSCT
- Alloimmunization due to repetitive transfusions (exchange of blood)
- Organ dysfunction due to SCA (possibly a reason why younger patients do better)
- Lack of matched sibling donors/ insurance.
Indications for performing allogeneic HSCT
- Stroke (most common and strongest indication to perform allogeneic HSCT.
- Abnormal transcranial doppler
- Acute chest syndrome
- Recurrent VOC not controlled with medical therapy or chronic transfusions
The complications with allogeneic HSCT:
- Transplant-related mortality approaches 7 to 10%, comparable with SCD-related mortality
- Graft rejection OR graft failure - less with myeloablative regimens (7 to 11%) compared to non-myeloablative regimens (11 to 50%)
- Graft-versus-host disease and related morbidity
- Transplant-related complications like lung injury, endocrine, and metabolic adverse events
The recent approvals of newer agents and the emergence of gene-editing techniques have expanded the options for SCA patients. Also, extending the benefit of HSCT to low-income countries remains a significant challenge.
Future Perspectives
Gene editing is a new therapy focus whereby researchers attempt to increase the HbF level in patients with SCA. This technique is being developed alongside HSCT. Many approaches to gene editing are in clinical trials right now.[26][27]
- Viral gene addition using lentivirus: The technique aims to add a modified beta or gamma-globin gene to reduce the HbS component and increase the HbA (beta-globin gene) or the HbF (gamma-globin gene).
- CRISPR (Clustered regularly interspaced short palindromic repeats): Targets the expression of BCL11A, which normally downregulates gamma-globin expression. By introducing insertions and deletions in the BCL11A erythroid lineage-specific enhancer on chromosome 2, BCL11A is downregulated, resulting in increased expression of the gamma-globin gene, which subsequently increases HbF.
Cost Factor
The annual cost of the voxelotor is approximately $125,000. Each vial of crizanlizumab costs approximately $2400, with a yearly cost of $84,852 and $113,136 per year for most patients. The monthly cost of the L-glutamine formulation is $3000 for adults and up to $1000 for the pediatric age group. A myeloablative regimen for HSCT can lead to a cost of approximately $280,000 at 100 days of care/admission.[28] In addition, the advanced level of expertise and dedicated infrastructure required to deliver such care also comes at a considerably high cost. Considering such high costs for the newer therapies, bringing them to lower-income regions like sub-Saharan Africa is a challenge, where approximately 6 million suffer from sickle cell anemia.
Prognosis
Most of the survival data in patients with SCA does not factor in the advent of new medications. The Cooperative Study of Sickle Cell Disease (CSSCD) (between 1978-88) reported the median age of death for women and men as 42 and 48 years, respectively. This study also showed that acute chest syndrome, renal failure, seizures, high leukocyte count, and low levels of HbF were associated with an increased risk of early death in patients with SCA.[29] More recent studies have shown that elevated tricuspid regurgitant jet velocity on echocardiography, prolonged QTc interval, pulmonary hypertension, high N-terminal pro-brain natriuretic peptide, history of asthma and/or wheezing, history of end-stage renal disease requiring dialysis, and the severity of hemolysis are independent risk factors towards early death in patients with SCA.[30]
More recent data combining nine studies from Europe and North America (evaluating 3257 patients) listed the following as predictors of mortality:
- Age (per 10-year increase in age)
- Tricuspid regurgitant jet velocity 2.5 m/s or more
- Reticulocyte count
- Log(N-terminal-pro-brain natriuretic peptide)
- Fetal hemoglobin[30]
With the approval of newer drugs (voxelotor and crizanlizumab) in 2019, increased use of hematopoietic stem cell transplant, and exploration of newer techniques like gene therapy, survival is bound to increase along with the quality of life.
Complications
SCA can lead to acute complications and chronic complications
Acute complications: Most acute complications are associated with occlusion of the small to medium-sized vessels (sometimes large-sized vessels) due to polymerization of HbS and hemolysis.
- Acute chest syndrome
- Sequestration crises: splenic or hepatic sequestration
- Fat embolism
- Bone infarction/necrosis
- Coagulopathy: increases the risk of both arterial and venous clots- stroke, myocardial infarction, venous thrombosis
- Ophthalmic: vitreous hemorrhage, retinal detachment, retinal artery/vein occlusion
- Aplastic crises: in association with parvovirus infection
- Papillary necrosis
Chronic Complications
- Delayed growth and development and growth retardation
- Cardiac: cardiomegaly, cardiomyopathy, left ventricular hypertrophy, arrhythmia, congestive heart failure
- Pulmonary: pulmonary edema, sickle cell lung disease, pulmonary hypertension
- Hepatobiliary: hepatomegaly, intrahepatic cholestasis, cholelithiasis, viral hepatitis
- Splenic complications: splenomegaly, hyposplenia, asplenia
- Renal: acute and chronic renal failure, pyelonephritis, renal medullary carcinoma
- Musculoskeletal: degenerative changes, osteomyelitis, septic arthritis, osteonecrosis, osteopenia/osteoporosis
- Neurologic: aneurysm, mental retardation
- Ophthalmic: proliferative sickle retinopathy, vitreous hemorrhage, retinal detachment, nonproliferative retinal changes
- Endocrine: primary hypogonadism, hypopituitarism, hypothalamic insufficiency
- Iron overload due to repeated transfusions and chronic hemolysis
Deterrence and Patient Education
SCA is a debilitating disease that affects a patient physically and has significant emotional and psychiatric consequences. The stigma of being diagnosed with SCA has been well documented. Many SCA patients are inaccurately labeled as drug seekers and opioid abusers due to the need for an inordinately high amount of opioids for pain control. In addition, frequent interactions with different providers (in the emergency rooms, hospital admissions, etc.) can lead to inconsistent care. In such a scenario, the patients need to be an advocate for themselves. The following points can act as a guide for patient education.
- Show consistency in outpatient clinics and show up for your appointments. Regularity in visits to your providers helps to build trust within the system.
- Discuss pain requirements for pain medications with your provider with an open mindset- They may appear restrictive in prescribing pain medications, especially opioids. Still, they are trying to help you by protecting you against overdosing.
- Use the same emergency room, or at least the ER within the same hospital system. It is useful and helps in developing familiarity with the people who work in that ER. It also allows easy access to your individualized plan of care, which your provider develops for such situations.
- Adherence to disease-modifying therapy will help reduce the events of pain crises and prevent long-term organ damage.
- Always be receptive to alternative ways of getting control over pain - including music therapy, self-hypnosis, and deep muscle relaxation.
- Patients can adopt protective measures- stay warm and avoid exposure to extreme temperatures, adequate hydration, and breathing exercises at home.
Enhancing Healthcare Team Outcomes
SCA is a systemic disorder that affects the entire body. The disease not only manifests with physical symptoms (pain crises, organ damage, etc.) but also has numerous psycho-social implications. Most patients with SCA belong to the African-American community and a minority to Hispanic and other communities, which makes them prone to certain prejudices. Besides, the high demand for opioids to manage chronic pain makes the situation even more challenging.[31] All providers must keep aside their inherent prejudice when caring for a patient with SCA, working collaboratively as an interprofessional team. Almost all specialties need to be involved in managing patients with SCA. However, the hematology team dedicated to taking care of SCA patients must be the primary physicians for these patients.
Specialties like ophthalmology, orthopedics, psychiatry, gastroenterology, and cardiovascular medicine interact closely with SCA patients. However, this does not diminish the importance of other specialties. Pharmacy and nursing also play a vital role. With the advent of newer drugs and infusions and SCA affecting liver and kidney function, pharmacists and nursing experts are required to ensure safe dosage and medication delivery to the patient.
The data presented here is derived mostly from large and small randomized clinical trials. [Level 1 and 2] Few aspects of care presented here are from cohort and case-control studies. [Level 3]