Continuing Education Activity
Hereditary spherocytosis, a type of congenital hemolytic anemia, is the most prevalent cause of hemolytic anemia due to an abnormal red cell membrane. These atypical erythrocytes are unable to maintain their normal biconcave shape due to genetic mutations in the membrane/cytoskeletal proteins that play a role in structural morphologic stability. The clinical manifestations vary according to the severity of the disease. This activity outlines the evaluation and management of hereditary spherocytosis and explains the role of the interprofessional team in providing quality care.
Objectives:
- Review the pathophysiology of hereditary spherocytosis.
- Describe the presentation of hereditary spherocytosis.
- Summarize the treatment of
hereditary spherocytosis.
- Identify the importance of improving care coordination among the interprofessional team in the prompt recognition of hereditary spherocytosis and referring these patients to a hematologist for better management.
Introduction
Hereditary spherocytosis (HS) is the most prevalent cause of hemolytic anemia due to an abnormal red cell membrane and classifies as a type of congenital hemolytic anemia. Oskar Minkowsky first described it in the early 1900s.[1] Erythrocytes are unable to maintain their normal biconcave shape due to genetic mutations in membrane/cytoskeletal proteins that play a role in structural morphologic stability. Deficient or abnormal proteins may include spectrin, ankyrin, band 3, and band 4.2, encoded by different genes.[2][3] The clinical manifestations vary based on the severity of disease and the type of genetic mutation.[1][2][3]
Etiology
Normal erythrocytes have a biconcave shape and possess relatively high plasticity. A lipid bilayer forms their membrane with proteins that adhere to the cytoskeleton and confer a normal biconcave shape providing optimal surface/volume relationship and adequate malleability, maintaining structural integrity and proper ion permeability.[1][4] With this shape, normal erythrocytes are able to buffer for small changes in osmolarity along with other changes in the intravascular environment.[4]
Hereditary spherocytosis results from a deficiency in one or more proteins involved in maintaining adequate adherence between the cytoskeleton and bilipid membrane. As the cellular surface is unable to be adequately supported by the cytoskeleton, the cell adopts an abnormal sphere shape that decreases surface to volume relationship. As the abnormal red blood cell membrane holds a higher tension, their structure is increasingly fragile and unable to buffer for several intravascular imbalances, such as a decrease in osmolarity.[1]
Spherocytes also have an abnormal ionic permeability that increases the influx of sodium to the intracellular space, which increases ATP requirements as the activity of the Na/K pump increases to compensate for the rise in intracellular sodium.[4]
Epidemiology
Hereditary spherocytosis is the most common genetic hemolytic disease. While it is more common in Northern Europe and North America, it may present in various racial populations. It is diagnosed in 1 out of every 2000 individuals, although a substantial number of asymptomatic cases may remain undiagnosed.[5]
Pathophysiology
Deficiency of RBC membrane proteins (mostly spectrin) results in defective linkage of RBC inner membrane cytoskeleton to its outer lipid bilayer. This defect results in loss of RBC membrane stability and deformability with progressive membrane lipid bilayer loss in the microvasculature. Membrane loss results in spherocytosis, a drop in mean corpuscular volume (MCV), an increase in mean corpuscular hemoglobin concentration (MCHC), and increased osmotic fragility of RBCs. Spherocytes are destroyed and cleared from circulation during passage through the spleen.[1][4]
Histopathology
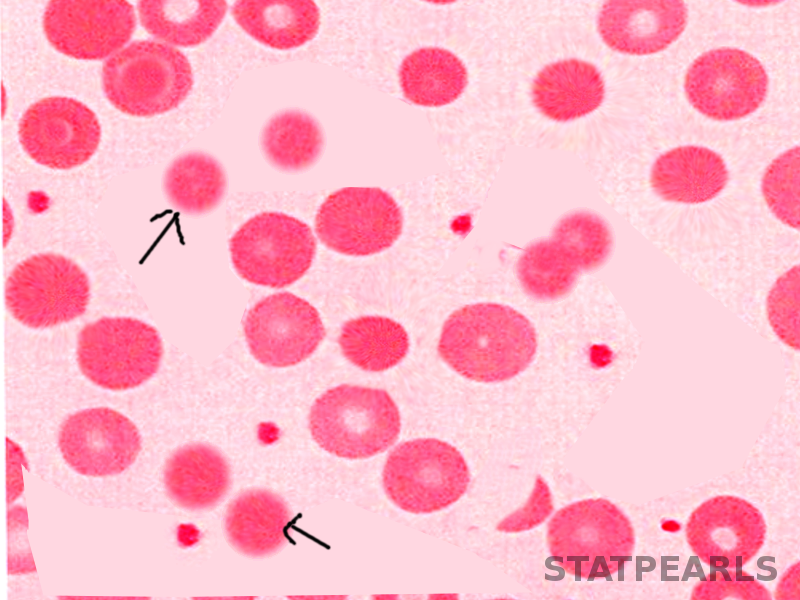
In microscopical blood smear evaluation, spherocytic red blood cells appear as smaller round cells lacking the normal central pale area due to the absence of a normal biconcave shape. Spherocytes may also be present in other types of hemolytic anemia, although more commonly in hereditary spherocytosis.[2][6]
Howell-Jolly bodies are frequently present in post-splenectomy blood smear microscopic evaluation; their absence after splenectomy may be a sign of residual functioning splenic tissue.[7]
History and Physical
Most patients with hereditary spherocytosis have a positive family history.[8] This disease should be suspected in any child presenting with splenomegaly, anemia, and hyperbilirubinemia. Patients may also present with asymptomatic cholelithiasis of undetermined etiology, commonly with a positive family history.[7]
The classic triad of jaundice, anemia, and splenomegaly is rare in neonates. The most common finding in this population is jaundice. Thus hereditary spherocytosis should be considered in neonates with hyperbilirubinemia of unknown cause. Additionally, spherocytes are seen less often in newborns along with several markers of the hemolysis that are typically present in older infants or adults.[9]
Most neonates with hereditary spherocytosis have a positive family history; this should increase suspicion and aid in the diagnostic workup of patients with an unknown cause of jaundice.[9]
Evaluation
Diagnostic basis revolves around history, clinical presentation, and laboratory results, which should include a blood smear, reticulocyte count, complete blood count, bilirubin level, and red cell osmotic fragility tests. Diagnosis is considered positive in patients who present Coombs-negative hemolysis with increased mean corpuscular hemoglobin concentration (MCHC), positive history for hereditary spherocytosis and spherocytes on microscopy, or by a positive result in a specialized test such as osmotic fragility, eosin-5-maleimide (EMA), or acidified glycerol lysis test (AGLT).[3][10]
Hereditary spherocytosis has a broad severity spectrum, and patients will have different clinical courses. The degree of anemia categorizes as severe for patients with hemoglobin lower than 8 g/dL, moderate for 8 to 10 g/dL, or mild with hemoglobin 10 to 11.5 g/dL in females, or 10 to 13.5 g/dL in males.[8]
Treatment / Management
Treatment centers on improving quality of life, avoiding the complications of hereditary spherocytosis, and treating them appropriately when present. For pediatric population hemoglobin is generally kept above 7 to 8g/dL; however, for newborns, there is no clear consensus regarding hemoglobin threshold to transfuse red blood cells. The decision to transfuse neonates focuses on their hemodynamic stability and degree of hemolysis.[1][7]
A previous study showed that erythropoietin treatment was beneficial for most infants with hereditary spherocytosis to increase hemoglobin levels and possibly decrease the need for red blood cell transfusion.[11]
Neonates with hyperbilirubinemia due to hereditary spherocytosis should be promptly treated to avoid kernicterus.[7] The decision to perform blood exchange transfusion therapy should result from the consideration of several factors including gestational and chronological age, and level of indirect bilirubin in addition to the rate of increase.[4][7]
Splenectomy largely improves symptomatology in most patients as the spleen is the principal organ where erythrocyte destruction takes place. However, while most patients show a positive outcome after surgical excision of the spleen, a study showed that clinical response to splenectomy correlates with the degree of spectrin deficiency.[12] The indication for splenectomy generally focuses on improving the quality of life. However, while the indication is clear for patients with severe hereditary spherocytosis, for patients with moderate disease there is no clear consensus and the decision for splenectomy in these patients is controversial.[1][7]
Due to the increase in erythropoietic activity compensating for hemolysis, patients with hereditary spherocytosis have increased consumption of folate. For this reason, adequate supplementation is necessary, particularly in patients with moderate to severe disease.[8]
Differential Diagnosis
The differential diagnosis for patients with hereditary spherocytosis is broad and includes other red blood cell disorders including elliptocytosis and its variants, hereditary stomatocytosis, pyropoikilocytosis, and ovalocytosis. Additionally, red blood cell enzymes disorders should also be considered, such as glucose-6-phosphate dehydrogenase (G6PD) deficiency or pyruvate kinase deficiency.[13][14]
Autoimmune hemolytic anemia (AIHA) is a common cause of hemolysis, and it commonly presents in adult patients. Unlike elliptocytosis, however, patients with AIHA commonly test positive for Coombs, and this disease does not commonly correlate with a positive family history.[15][16]
Prognosis
The overall prognosis for patients with hereditary spherocytosis is considered good. However, it may vary depending on the disease severity.
Complications
Approximately 50% of patients with hereditary spherocytosis present with anemia, and 10 to 15% with splenomegaly or jaundice.[17] The majority of patients will present partially compensating hemolysis in addition to mild anemia.[7][17]
Patients who undergo splenectomy are at risk of sepsis. The most common causing organisms are encapsulated and include Streptococcus pneumoniae with the highest incidence, Neisseria meningitides, and Haemophilus influenzae. The risk of sepsis is associated with the age of the patient.[7]
Cholelithiasis is one of the most common complications and usually presents in patients between the ages of 10 and 30 years, although younger patients may also present with cholelithiasis.[18]
Some patients may also present with hematologic crises, which may be hemolytic, aplastic, or megaloblastic.[19]Hemolytic crisis is the most common of these and presents with fever; it may be triggered by viral infections, although it may present spontaneously.[19]
Aplastic crises are generally worse than hemolytic crises and may be the cause of acute heart failure; it has correlations with parvovirus B19. Children commonly present with chills and fever, abdominal pain, myalgias, and generalized maculopapular rash. The clinical course of aplastic crisis usually starts with a decrease in hemoglobin and reticulocytes, followed by a reduction in bone marrow erythroblasts and bilirubin. During recovery neutrophils and platelets increase first, then subsequently by reticulocytosis and gradual increase in hemoglobin levels.[20]
Megaloblastic crises are infrequent and usually caused by a deficiency in folate levels. These complications are treatable with adequate folate supplementation.[7]
Deterrence and Patient Education
Patients with hereditary spherocytosis, especially parents of younger children will benefit from comprehensive educational supplementation. These patients will require periodical follow up, and monitoring. Ideally, they should be able to identify early signs and symptoms of disease complications such as aplastic crises or cholelithiasis.[7]
Affected patients who undergo splenectomy will require additional prophylactic vaccination against encapsulated organisms and follow-up to monitor for complications.[7]
For patients who have moderate disease, the decision to perform splenectomy is controversial and extensive counseling, and educational plan are warranted before drawing a conclusion, there should be a consensus should between the patient, family, and physicians.[7]
Pearls and Other Issues
Complications:
- Aplastic crisis secondary to parvovirus B19 infection
- Pigment gallstones more frequent in the second and third decades of life and may result in obstructive jaundice
- 3. Gout, ulcer, and growth retardation (less common)
Enhancing Healthcare Team Outcomes
Neonatal patients with high bilirubin levels should be promptly treated to avoid kernicterus. Diagnostic workup should follow to determine the cause of hyperbilirubinemia, and hereditary spherocytosis should be a consideration as newborns do not typically present the same signs and symptoms as older infants.
As discussed above, the indication for splenectomy should focus on improving the quality of life. Particularly for patients with frequent aplastic crises, a decision should include a thorough discussion with patients or parents regarding the risks and benefits of surgical therapy.[2]
Healthcare workers, including physicians, PAs, nurse practitioners, nurses, and pharmacists who see patients with anemia in young people should consider the possibility of hereditary spherocytosis and refer these patients to a hematologist for diagnostic workup. Even after hematologic referral, an interprofessional team approach including all of the above disciplines working collaboratively can help achieve the best patient outcomes, regardless of the therapeutic direction chosen in any specific case.