Continuing Education Activity
Tay-Sachs disease is a rare autosomal recessive progressive neurodegenerative disorder. Patients usually exhibit a severe clinical course with death in early childhood. Early diagnosis of Tay Sachs is clinically very challenging because of the subtle clinical features and nonspecific biochemical markers. Accurate diagnosis is important both for proper management and to reduce morbidity and also for parental counseling and prenatal diagnosis. This activity reviews the pathophysiology, clinical features, evaluation, and treatment of Tay-Sachs disease; and highlights the role of the interprofessional team in the management.
Objectives:
- Describe the etiology of Tay-Sachs disease.
- Outline the evaluation of a patient with Tay-Sachs disease.
- Outline the management options available for Tay-Sachs disease.
Introduction
Tay Sachs disease (TSD) is a progressive, lethal neurodegenerative disorder caused by a deficiency of enzyme hexosaminidase-A resulting in the accumulation of GM2 gangliosides. Based on the presentation age, the disease is classified into infantile, juvenile, and adult forms. Early diagnosis of Tay Sachs is clinically challenging because of subtle clinical features and nonspecific biochemical findings. Accurate diagnosis is important both for proper management and to reduce complications associated with the disease.
Etiology
Tay Sachs disease is an autosomal recessive disorder caused by a mutation in the gene HEXA, which encodes the enzymes beta-hexosaminidase A. HEXA is located at 15q23. More than 130 mutations have been identified so far, and it includes single gene deletions, substitution, insertion splicing alteration, duplication, and complex gene rearrangements.[1]
Epidemiology
Tay Sachs disease is rare in the general population, and the incidence is about 1 in 320,000 live births in the United States, whereas the carrier frequency is about 1 in 250.[2] The disease is more frequent in the people of Ashkenazi Jewish heritage, i.e., those of central or eastern European descent. Epidemiological studies on carrier state in the Jewish community in the US showed a prevalence of about 1 in 29, and 1 in 3500 live birth is affected.[3] A Cajun community of Lousiana, an old order Amish community in Pennsylvania, and non-Jewish French Canadians living near St. Lawrence also have a high incidence of TSD.[4]
Pathophysiology
Tay Sachs disease is caused by the deficiency of the enzyme Beta hexosaminidase A (Hex A), which is responsible for GM2 ganglioside degradation. Alpha and beta subunits of Hex A are synthesized at the endoplasmic reticulum. Following glycosylation, intramolecular disulfide bond formation, and dimerization in the endoplasmic reticulum, the enzyme is transported to the Golgi network. Post-translational modification of the enzyme with mannose-6-Phosphate is the most important step, which helps the lysosome to recognize the enzyme. The presentation of GM2 ganglioside to the active site of Hex A requires an activator protein GM2A, which makes the enzyme lipophilic.[5][6]
Gangliosides are the major glycolipid of the neuronal cell membrane, ensuring normal cellular activity.[7] Ganglioside expression in the brain is highly region-specific and is highly regulated and correlated with neurodevelopmental milestones, including neural tube formation, neuritogenesis, axonogenesis, synaptogenesis, and myelination. Ganglioside accretion occurs early as the 10th week of gestation and continues through five years of life. It also plays a major role in modulating ion channel function and receptor signaling, ensuring optimum function and adaption of neuronal circuits involved in neurotransmission, memory, and learning. But deficiency of Hex A causes accumulation of the gangliosides to toxic levels, especially in the neurons. While it is clear that the accumulation of ganglioside is the cause, the exact mechanism that translates the primary insult into neuronal death is unclear.[8] Progressive neurodegeneration, microglia proliferation, and accumulation of the complex lipids in neuronal macrophages occur.[8]
Other pathological processes in the disease process include abnormal endosomal transport, impaired autophagy, progressive accumulation of alpha-synuclein, and anti-ganglioside antibodies.[9]
Histopathology
Hexaminidase levels are virtually absent from all the tissues, including plasma and leucocytes. Gangliosides accumulation is seen in all tissues but predominately seen in the central and autonomic nervous system and the retina. Patients with acute infantile Tay Sachs have an excessive accumulation of GM2 ganglioside (at least <12% of dry weight). In contrast, the adult forms have less accumulation, and even it may be restricted to specific regions. For example, the cortex is almost unaffected in adult forms, whereas brainstem nuclei, spinal cord, and hippocampus are markedly affected.[10][11]
On histologic examination, neurons are ballooned with cytoplasmic vacuoles, which constitute distended lysosomes with ganglioside. Sudan Black B and oil red O are typically positive.[12] Under electron microscopy, the most prominent type seen is a whorled configuration within lysosomes resembling onion-skin layers.
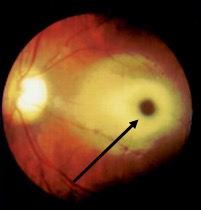
GM2 gangliosides accumulate in the retinal ganglion cells, particularly in the margins of the macula, causing cherry-red spots. The ganglion cells are in high density around the fovea, swelling of which causes a grey-white appearance, whereas the center of the fovea does not have ganglion cells, and those retain the reddish appearance. Toxic accumulation of intra-lysosomal gangliosides causes early visual symptoms.[12]
History and Physical
Tay Sachs disease manifests in a wide spectrum, and it has infantile, juvenile, and adult-onset types. Infantile Tay Sachs is a prototype of degenerative diseases of grey matter in infancy. The infant is usually normal at birth, and the symptoms start between 3 to 6 months but can manifest as early as one week of life.[7] Nonhydrops fetalis can be the initial presentation representing prenatal involvement. The classical initial clinical features are mild motor weakness, irritability, and hypersensitivity to auditory and other sensory stimuli. An exaggerated startle (auditory myoclonus) response is considered an initial and helpful sign in the diagnosis.
A cherry-red spot in the retina is almost a near-specific finding in Tay Sachs patients by fundoscopic examination(Figure.1).[13][14] This finding has been noted as early as two days of life. The cherry red spot is due to attenuation of the normal color of the macula and choroid contrasted with pallor produced by the swollen ganglion cells in the remainder of the retina. By six months of age, all the patients develop this finding, and by 12 to 18 months of age, vision is reduced, and by 30 months, most patients become blind. Patients also develop narrowing of retinal vessels, nystagmus, and optic atrophy.[15] The child also has doll-like facies.
Neurological symptoms are the hallmark of Tay Sachs disease. Infants are usually hypotonic since birth and present with developmental delays or regression by four to six months of age. By eight to ten months, symptoms rapidly progress, and spontaneous and voluntary movements diminish, and the infant becomes progressively less responsive.[16][17] The patient also develops seizures by twelve months, with the most common type being the tonic-myoclonic type. Spasticity and seizures mark the final phase of the illness. Myoclonic seizures can be massive and multiple and often refractory to medications (more than two-thirds of patients needed more than two anticonvulsants).[18] Generalized, focal, and gelastic seizures also can occur. Around the same age, the patient also develops ataxia, dyskinesia, sleep disturbances, episodes of screaming, and irritability.
By 18 months of age, patients usually develop macrocephaly.[19] Increasing head circumference is due to reactive cerebral gliosis and is not due to hydrocephalus. By two years of age, patients deteriorate and develop decerebrate posturing, dysphagia, unresponsive and vegetative state.
Cardiovascular complications are rare but can occur due to the accumulation of the substrate. Prolonged QT interval and nonspecific T wave changes are reported.[20] Hepatosplenomegaly is typically absent in Tay Sachs disease. Patients often develop infections, and respiratory infections are reported frequently as the cause of death. Interestingly, Tay Sachs carriers are more resistant to Mycobacterium infection, which typically lack alpha units and alpha-beta heterodimer of beta-hexosaminidase, and have increased production of HexB (beta subunit) by as much as 200%. The beta subunit of HexB confers immunity against Mycobacterium.[21]
Juvenile Tay Sachs disease manifests in early childhood between ages two to ten, caused by the reduced activity level of Hex A. The earlier the onset of the symptoms, the more quickly the disease progress. Early symptoms include incoordination, clumsiness, and muscle weakness. Other common symptoms include ataxia, dysarthria, dysphagia, and the progression of spasticity.[22] A cherry red spot is not consistently observed. Optic atrophy and retinitis pigmentosa can be observed later in life.[23] A vegetative state with decerebrate posturing occurs by age 10 to 15 and is followed within a few years by death, usually from a respiratory infection.
Adult Tay Sachs disease is an infrequent disorder, and diagnosis is often delayed at least by eight years of age. The adult form is less aggressive, resulting from a small mutation and high residual activity of Hex A, which is at least 5% to 20% of normal activity. Typical symptoms start in adolescence or early adulthood but can appear later(20 to 30 years of life).[24] Affected individuals have several different phenotypes, including progressive motor neuron disease, dystonia, and spinocerebellar degeneration. Mentation and verbal skills are affected later in the course of illness. Psychiatric features are also very common in adult-onset Tay-Sachs disease. About 40% of individuals develop psychiatric manifestations without dementia, including recurrent psychotic depression, hebephrenic schizophrenia with disorganization of thoughts, delusion and hallucination, paranoia, and bipolar symptoms.[25][26] In any patient with psychiatric illness, the presence of cognitive decline or neurological symptoms should raise suspicion of storage disorders.
Evaluation
The classical clinical findings of progressive weakness with developmental delay or regression, inattentiveness, and exaggerated startle response with physical findings of a cherry-red spot, generalized hypotonia with sustained clonus, or hyperreflexia should warrant further evaluation for Gangliosidosis. The first step in the evaluation involves the demonstration of Hex A and total Hexosaminidase levels in the serum. Individuals with infantile form will have no or extremely low level of enzyme activity (0-5%) in addition to normal or elevated levels of Beta hexosaminidase levels (HEX B isoenzyme). Individuals with juvenile or adult forms will have low enzyme activity (10 to 15%). If the initial testing shows reduced enzyme activity and if the patient is from Azheknazi or French Canadian background, consider targeted gene testing.[27]
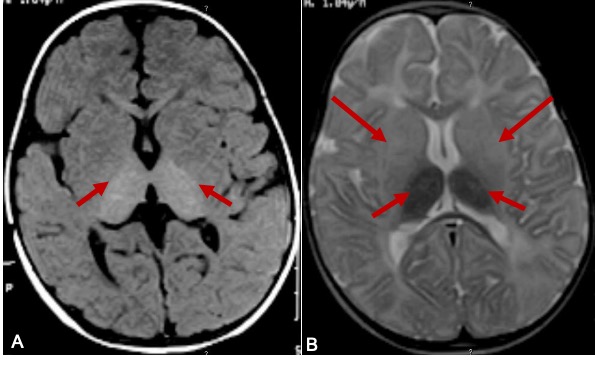
Neuroradiological findings in Tay Sachs disease are described in three phases of the clinical course.[28][29] In the initial phase, cerebral white matter and basal ganglia show hypodensity in CT and T2 hyperintense signal changes on MRI (Figure.2). Caudate nuclei are enlarged and extend into the lateral ventricle in the first and second phases. The brain becomes atrophic in the final phase. The combination of hyperdensity on CT images and T2W hypointense/ T1W hyperintense MRI signal involving bilateral thalami is suggestive of Tay Sachs disease(Figure 2).[30] MR spectroscopy demonstrated an increase in myoinositol/creatinine and choline/creatinine ratio with a decrease in N-acetyl aspartate/creatinine ratio. MR spectroscopy is very sensitive and specific to neuroaxonal damage in late-onset Tay-Sachs disease.[31] Especially MR spectroscopy might be a good tool to detect and quantify the neuronal damage and to monitor the response to treatment in the late-onset Tay Sachs disease patients on pharmacological agents to reduce ganglioside accumulation
For carrier testing in Ashkenazi Jew ancestry, the first step depends on whether comprehensive Ashkenazi Jewish carrier screening is desired or only Tay Sachs screening is required.[32] Comprehensive Ashkenazi Jewish carrier panel includes Tay Sachs, Canavan disease, familial dysautonomia, Gauchers, Bloom disease, Niemann Pick type A and B, Mucolipidosis IV, and Fanconi anemia type C.
Molecular genetic testing includes sequencing, targeted analysis for pathogenic variants, and deletion/duplication analysis. The targeted analysis is done if the enzyme activity is absent or low on the initial assay. The panel consists of six common pathogenic variants. The panel includes three null alleles p.Tyr427IlefsTer5, c.1421+1G>C, and c.1073+G>A, which in the homozygous or compound heterozygous state is associated with Tay Sachs. Allele p.Gly269Ser is associated with an adult-onset form of Hex A deficiency. Two pseudo deficiency alleles ( p.Arg247Trp and p.Arg249Trp ) are not associated with neurological disease but with reduced degradation of the synthetic substrate when Hex A activity is determined in-vitro.[33] The test results need to be interpreted carefully because the pseudo deficiency allele does not reduce the enzyme activity with the natural substrate in vivo. About 35 % of non-Jewish individuals identified as heterozygotes by Hex A enzyme testing are a carrier for the pseudo deficiency allele, whereas it is 2% in the Jewish population.
Prenatal testing on fetal cells can be done by chorionic villus sampling at 10 to 12 weeks of gestation or by amniocentesis at 15 to 18 weeks of gestation in families when Hex A enzyme assay shows parents are heterozygous and molecular genetic testing has ruled out pseudo deficiency allele in either parent. In families with identified pathogenic variants, preimplantation genetic testing is an option.
Treatment / Management
Treatment of Tay Sach disease is largely supportive and aims at providing adequate nutrition, controlling seizures, managing the infectious disease, protecting the airway, and early aggressive physical and occupational therapy. Seizure control usually needs multiple antiepileptics. However, seizure becomes progressive and change in pattern, so frequent dose change and new medications are important. As the child with Tay Sachs disease becomes more disabled and debilitated, good bowel management becomes essential.
Therapeutic modalities for Tay Sachs disease include enzyme replacement therapy, cell transplantation, substrate reduction therapy, enzyme enhancing therapy, and gene therapy.
Enzyme replacement therapy (ERT) is a promising option for Tay Sach disease. Currently, ERT is less effective in Tay Sachs disease due to the inability to cross the blood-brain barrier and prevent neurological complications. Another major challenge in ERT is to synthesize both of the subunits. The synthetic Hexa A is treated with alpha-mannosidase to expose mannose 6 residues on the N-Glycans.[34]
Enzyme enhancing therapy: Most mutations causing Tay Sachs are not localized to the active site but often cause instability of native folded protein. The strategies to reduce the substrate includes the use of molecules called chaperones to stabilize the enzyme. Interestingly the pharmacological chaperones used are enzyme-specific competitive inhibitors. A trial of Hexa A inhibitor pyrimethamine has shown to increase the Hex A levels to 4 folds, but clinical benefits are not reported.
Substrate reduction therapy (SRT): The rationale behind SRT is balancing substrate synthesis with that of diminished enzyme degradative power. Substrate reduction drug Miglustat (N- butyldeoxynojirimycin) has been successful in mouse models but not in humans. Currently, the FDA did not approve the use of Miglustat in Tay Sachs.
Gene therapy: Since Tay Sachs is caused by a single gene disorder will be an excellent option for gene therapy. Great progress has been achieved so far by developing adeno associated virus-based vectors. The major limitation of adeno based vectors is their capacity to carry constructs. For clinically significant therapy, a vector should carry Hex A isoenzyme that should carry both alpha and beta subunits. Genome editing therapies using zinc-finger nucleases are being explored. There were few scattered reports of tumor development in mice models kept alive for a prolonged time following gene therapy, so a cautious approach is warranted. Huge efforts are being invested in developing viral vectors for the effective delivery of isoenzymes.[34]
Bone marrow transplantation (BMT): The transplantation of ex vivo modified multipotent neural cells with human HEX A gene expression produced by retroviral transduction is another major breakthrough approach.
Overall, adequate production and distribution of Hex A are required for a better therapeutic effect in Tay Sachs disease. So far substrate reduction therapy, bone marrow transplantation, and enzyme replacement therapy have shown low efficacy in preventing Neurodegeneration. Hence, a combination of multiple therapies at an early age is important since myelination defects appear early and worsen with time.
Differential Diagnosis
Activator deficient Tay Sachs disease: A variant of Tay Sachs disease with classical findings of neuro regression, exaggerated startle response, and cherry red spot of the macula without hepatosplenomegaly. This condition should be considered in patients with classical findings of Tay Sachs but with normal Hex A and Hex B levels. Ganglioside accumulation occurs due to the deficiency of GM2 glycoprotein( intralysosomal glycoprotein) required for GM2 Ganglioside degradation.[35]
Sandhoff disease is characterized by progressive neurodegeneration starting at 6 months of life associated with hyperacusis, cherry red spot, and blindness. Sandhoff disease is indistinguishable from Tay Sachs in clinical course, diagnosis, and management except for the visceral and bone involvement. Hepatomegaly is common in Sandhoff disease. Sandhoff is a severe form of Tay Sachs disease caused by a mutation in the Hexosamidianse B (Hex B) gene. It is usually not limited to any specific ethnic group.
Progressive neurodegeneration with cherry-red spot is also common in other lysosomal storage disorders, including GM1 Gangliosidosis, Sandhoff disease, infantile Gaucher's disease, Niemann Pick disease type A, and galactosialidosis.
The differential diagnosis for late-onset Tay Sachs disease with neurological findings includes adolescent-onset Spinal muscular atrophy, Friedreich ataxia, amyotrophic lateral sclerosis, and Kuf's disease ( adult-onset neuronal cerebral lipofuscinosis) and late-onset forms of lysosomal storage disorders. Differential diagnosis of adult-onset Tay Sachs with neuropsychiatric manifestation includes Hepatolenticular degeneration, Niemann Pick Type C, cerebrotendinous xanthomatosis, ceroid neuronal lipofuscinosis, metachromatic leucodystrophy, and X-linked adrenoleukodystrophy. All the above diagnoses should be considered in young patients with refractory or unusual neuropsychiatric symptom clusters.
Prognosis
Tay Sachs disease is a progressive neurodegenerative disease. There is progressive neurological deterioration, and the seizures often remain refractory to treatment. Even with the best care, patients with infantile Tay Sach's disease usually die by age of 4 to 5 years. Death usually results from recurrent infections.
In late-onset disease, there are progressive gait difficulties and motor impairment, which often requires the use of adaptive equipment and mobility assistance. Besides, psychiatric symptoms often remain resistant to treatment. Progressive neurological deterioration often leads to a vegetative state, and death usually occurs by 10 to 15 years of age.
Complications
The complications include progressive neurological deterioration, spasticity, refractory seizures, and progressive visual impairment, finally leading to a vegetative state.
Late-onset disease patients develop progressive motor impairment and balance issues and are at risk of falls. They are also at risk of developing psychiatric symptoms, which may remain resistant to treatment.
Consultations
Following the initial diagnosis, both infantile-onset and late-onset Tay Sach's disease patients require consultations with multiple professionals. Evaluation by a neurologist is essential to assess and manage the neurologic symptoms. This includes brain MRI and EEG and assessing the need for antiepileptic drug treatment and monitoring. Ophthalmology evaluation is required to assess visual impairment and its progression. A speech therapy referral is required to assess the swallowing dysfunction and risk of aspiration. In case of risk of aspiration referral for gastrostomy and involvement by feeding, the team is required. Respiratory team involvement is essential to assess the airway. Physiotherapy and occupational therapy referrals are required to manage neuromuscular impairment. Referral to clinical genetics services is required for genetic counseling, screening of at-risk family members, and prenatal or preimplantation genetic diagnosis. Patients with late-onset Tay Sach disease might require referral to a psychiatrist or psychological medicine team for the assessment of psychiatric symptoms and the need for treatment. It is also important to provide appropriate social support to the family through the social work team's involvement.
Deterrence and Patient Education
Parents and caregivers should be appropriately counseled regarding the diagnosis, progression, and anticipated complications of Tay Sach disease. Families should be informed about the expected outcome, such as progressive neurological deterioration, the refractory nature of the seizures, and the risk of aspiration and recurrent infections. Late-onset Tay Sach's disease (Juvenile and adult-onset) patients should be informed about the risk of falls due to ataxia, and appropriate measures such as assistive devices should be advised. In addition, it is also difficult to treat psychiatric symptoms.
Appropriate genetic counseling should be offered to those who are carriers and at risk of being carriers. Autosomal recessive disorders result when one copy of the abnormal gene for the same trait is inherited from each parent. Thus, parents of an affected child with Tay-Sach disease are obligate heterozygote carriers. Thus, two carriers (father and mother) have a 25% chance that the offspring is affected and a 25% chance that their offspring is healthy, while half of their offspring (50%) may be carriers exactly like the parents. Heterozygotes or carriers are usually asymptomatic.
Enhancing Healthcare Team Outcomes
Tay Sachs disease has no curative treatment, and it is a progressive neurodegenerative disease. The newborn screening for Tay Sachs is not universally available currently. Early recognition of the disease is clinically very challenging, especially with the juvenile-onset and late-onset forms. A high index of suspicion is important when evaluating a patient from a high-risk group. Since Tay Sachs disease is a prototype of degenerative disease of the grey matter of infancy, any infant with developmental delay or rapid regression Tay Sachs disease should be considered in the differential diagnosis. In addition, the onset of psychiatric symptoms in clusters with neurological findings should prompt further testing for adult-onset Tay Sachs disease.
Tay Sachs disease is managed by a multidisciplinary team approach, including a neonatologist, pediatric neurologist, geneticist, nutritionist, physiotherapist, and occupational therapist. Genetic counselors play a significant role in providing parental reproductive risk counseling and prenatal diagnosis. Tay Sachs families need medical, educational, psychosocial, and financial support services in their local communities.