Continuing Education Activity
Mastocytosis is a disorder characterized by mast cell accumulation, commonly in the skin, bone marrow, gastrointestinal tract, liver, spleen, and lymphatic tissues. The World Health Organization (WHO) divides cutaneous mastocytosis into 3 main categories. The first involves 3 or fewer lesions called mastocytomas. The second involves between 4 and 100 lesions and is referred to as urticaria pigmentosa. The third category involves diffuse cutaneous involvement. Urticaria pigmentosa is the most common cutaneous mastocytosis in children and can present in adults as well. It is generally considered a benign, self-resolving condition that often remits in adolescence. Unlike the forms of mastocytosis that are solely seen in adults, internal organ involvement is rare in urticaria pigmentosa. This activity describes the risk factors, evaluation, and management of urticaria pigmentosa and highlights the role of the interprofessional team in enhancing care delivery for affected patients.
Objectives:
Differentiate urticaria pigmentosa from other skin disorders with similar presentations to ensure accurate diagnosis and appropriate management.
Assess the components of an appropriate evaluation for a patient with urticaria pigmentosa.
Identify management strategies for a patient with urticaria pigmentosa.
Collaborate amongst an interprofessional team involved in managing patients with urticaria pigmentosa to ensure a multidisciplinary approach and to enhance prompt and thorough delivery of care.
Introduction
Mastocytosis is a disorder characterized by mast cell accumulation, commonly in the skin, bone marrow, gastrointestinal (GI) tract, liver, spleen, and lymphatic tissues. The World Health Organization (WHO) divides cutaneous mastocytosis into 3 main presentations. The first has solitary or few (≤3) lesions called "mastocytomas." The second, urticaria pigmentosa (UP), involves multiple lesions ranging from >10 to <100 lesions. The last presentation involves diffuse cutaneous involvement.
UP is the most common cutaneous mastocytosis in children, but it can form in adults as well. It is considered a benign, self-resolving condition that often remits in adolescence. Unlike adult forms of mastocytosis, there is rarely any internal organ involvement in UP.[1]
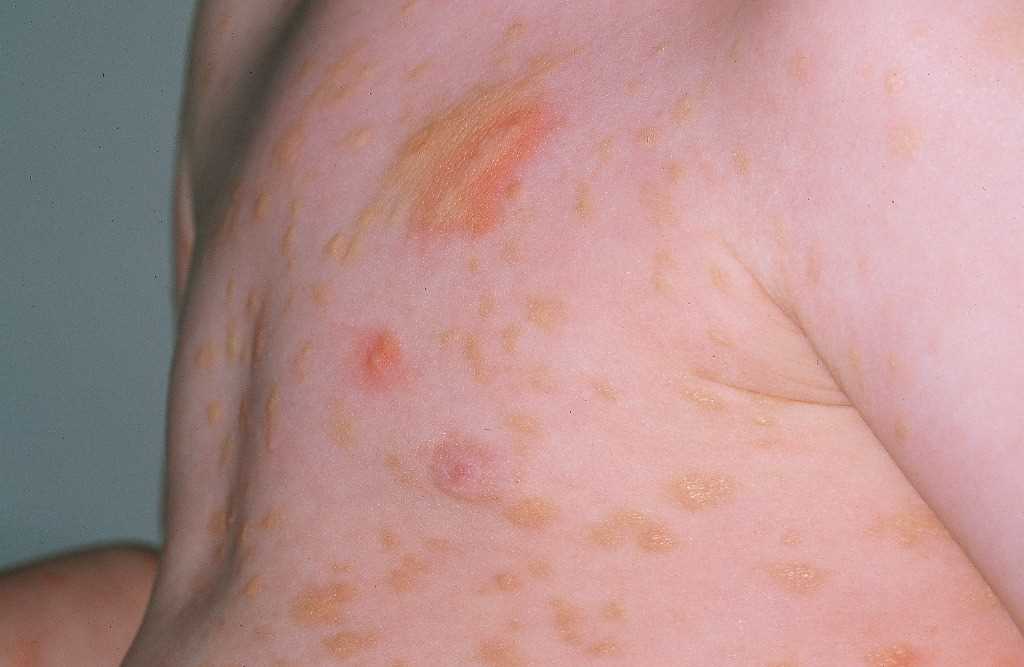
What makes UP particularly distinctive is its tendency to manifest as small, itchy, reddish-brown, or yellowish-brown spots or lesions on the skin, commonly referred to as "urticaria" or hives. These spots typically appear in childhood and can persist throughout a person's life. The condition can vary in severity, and while it is often benign, it may sometimes cause symptoms and complications related to mast cell activation. Understanding the features, causes, and management of UP is essential for healthcare professionals.[2]
Etiology
UP is caused by several activating mutations in the KIT gene. When exposed to certain triggers, mast cells release mediators that cause the symptoms of mastocytosis. The released mediators are histamine, eicosanoids, prostaglandins, leukotrienes, heparin, proteases, and cytokines. Triggers include certain foods, exercise, heat, Hymenoptera and venomous stings, local trauma to skin lesions, alcohol, narcotics, salicylates and other nonsteroidal anti-inflammatory drugs (NSAIDs), polymyxin B, and anticholinergic medications. Some systemic anesthetic agents may induce anaphylaxis.[3]
Epidemiology
Mastocytosis can present at birth or develop at any time into late adulthood. It occurs in all races and has no gender preferences. Although most patients do not have a family history of mastocytosis, familial cases have been reported.[4] Cutaneous mastocytomas occur in 15% to 50% of patients, while UP occurs in 45% to 75%, and diffuse cutaneous involvement occurs in less than 5% to 10% of mastocytosis patients. Most reported cases are in whites. The cutaneous lesions of most types of mastocytosis are less visible in persons with more heavily pigmented skin. The apparent prevalence of autism spectrum disorders in children with mastocytosis was found to be 10 times higher than in the general population.[5]
Pathophysiology
Mast cell precursors express CD34, the tyrosine kinase receptor KIT (CD117), and IgG receptors (Fc gamma RII). KIT can be activated by its ligand, stem cell factor (SCF), which induces mast cell growth and maturation and prevents apoptosis. Bone marrow stromal cells, keratinocytes, endothelial cells, fibroblasts, and reproductive Sertoli and granulosa cells produce SCF. The pathogenesis of mastocytosis is due to several mutations in KIT. The most common is an activating mutation in codon 816, where there is a substitution of the amino acid aspartic acid (D) with valine (V, ie, D816V) or another amino acid. These amino acids result in constitutive ligand-independent activation of the receptor. Other additional pathogenic factors exist that still need more research.[6][7] The increased concentrations of soluble mast cell growth factor in lesions of cutaneous mastocytosis are believed to stimulate mast cell proliferation, melanocyte induction, and melanin pigment production. The induction of melanocytes explains the hyperpigmentation that is commonly seen in cutaneous mast cell lesions. The stimulation of pruritus reported in mastocytosis is associated with the production of interleukin (IL)–31.[8] Impaired mast cell apoptosis has been postulated to be involved, as evidenced by the up-regulation of the apoptosis-preventing protein BCL-2 demonstrated in the patients.[9] IL-6 levels are elevated and correlated with disease severity, indicating the involvement of IL-6 in the pathophysiology of mastocytosis.[10]
Histopathology
Diagnosis of UP can be made clinically. However, a definitive diagnosis requires a skin biopsy. An anesthetic agent should be injected without epinephrine adjacent to, not directly into, the lesion chosen as the biopsy specimen to avoid mast cell degranulation, which makes histologic examination difficult.
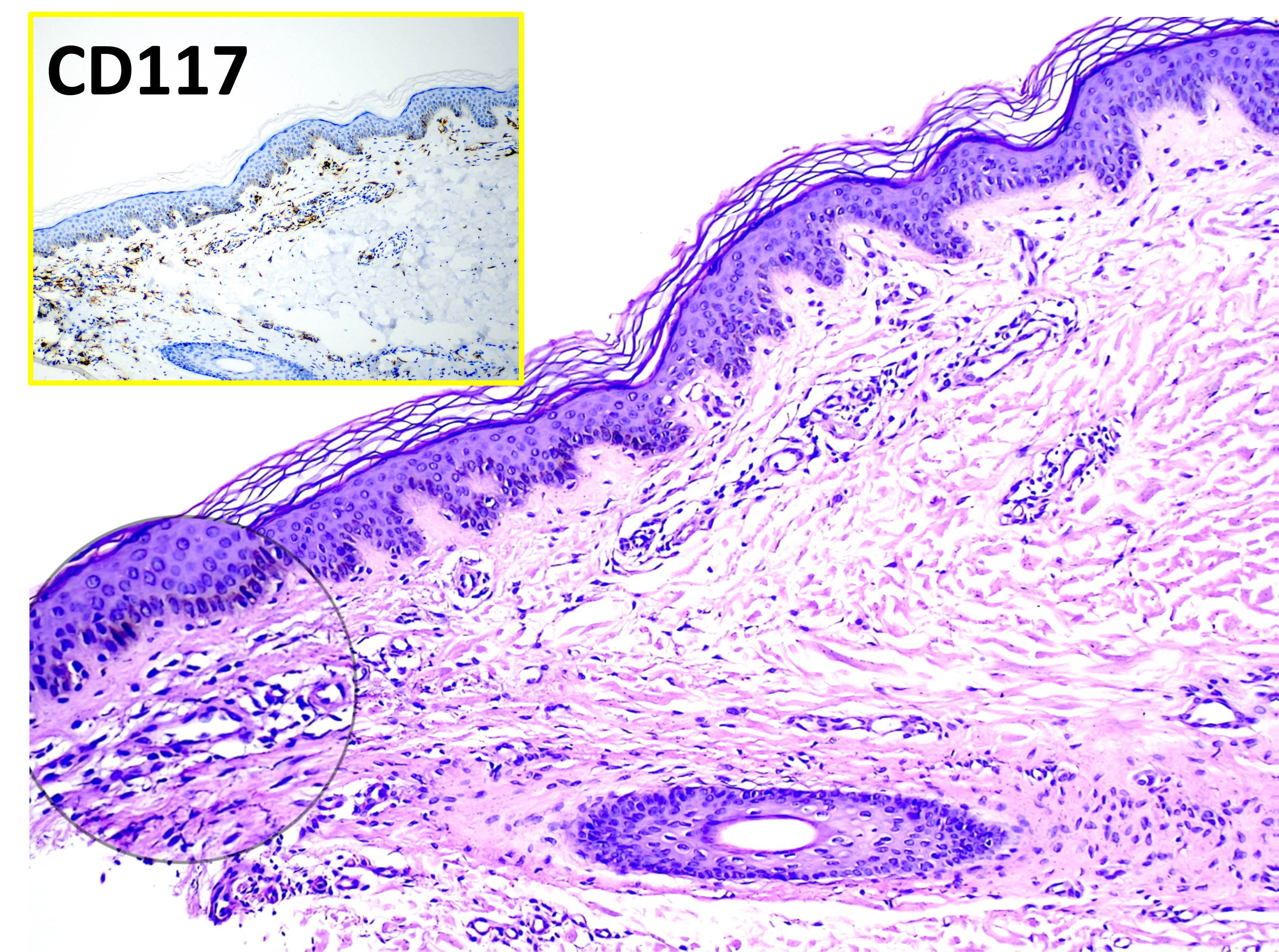
The hallmark finding histologically for UP is having an increased amount of mast cells in the dermis. UP skin lesions can have a 40-fold higher mast cell count than normal skin.[11] Mast cells have a rounded or cuboidal appearance (see Image. Mastocytosis). Mast cell nuclei are rounded with surrounding ample cytoplasm, producing a “fried egg” appearance. The typical presentation of mast cells is a single nucleus, yet there have also been atypical histological findings reported in 4 patients with UP that showed mast cells with bilobed and multilobed morphology.[12] In nodular UP, mast cells are observed in dense aggregates and may extend through the entire dermis and into subcutaneous tissue.
If the lesion from which the biopsy specimen was taken was traumatized during harvest, edema and eosinophil infiltrates may be present. The hyperpigmentation of cutaneous mastocytosis is secondary to increased melanin in the basal cell layer and melanophages in the upper dermis. Stains that help identify mast cells include toluidine blue, Leder, Giemsa, tryptase, and CD117 (KIT). Biopsy specimens from normal skin in patients with mastocytosis will not show an increased amount of mast cells. Therefore, if there are no skin lesions and systemic mastocytosis is suspected, a bone marrow biopsy or biopsy from the GI tract may be needed. A total serum tryptase level can help identify the extent of mast cell disease. A tryptase level >20 ng/ml represents 1 of the minor criteria for systemic mastocytosis.[13]
History and Physical
UP has small, monomorphic tan to brown macules or papules distributed mainly on the trunk and classically spares the central face, palms, and soles (see Image. Urticaria Pigmentosa). It is similar to the lesions typically observed in adults. Lesions can be few or numerous and are usually about 1 to 2 cm in size; however, they can be larger. Darier’s sign is the formation of a wheal upon stroking or rubbing skin lesions and suggests the diagnosis of cutaneous mastocytosis. The wheal forms due to the release of mast cell mediators, which can contribute to the systemic symptoms that may occur after stroking the lesion. Some lesions may blister after stroking the lesion. Darier’s sign is more pronounced in children than in adults due to an increased density of mast cells in lesions of children. In approximately half of the patients, stroking macroscopically uninvolved skin produces dermographia. Large hemorrhagic bullous and infiltrative small vesicular variants occur. Blistering predominates in infancy, whereas the grain-leather appearance of the skin and pseudoxanthomatous presentation develop with time. Anaphylactic shock may occur.
Systemic symptoms can occur, such as pruritus, abdominal pain, flushing, diarrhea, dizziness, palpitations, and syncope. Twenty-five percent of the patients with UP experience gastrointestinal symptoms. Complaints of fever, malaise, night sweats, bone pain, epigastric distress, weight loss, and problems with cognitive disorganization often signal the presence of extracutaneous disease. Death can even occur from the extensive mast cell mediator release.[14]
Evaluation
If the patient complains of GI symptoms, a barium study or endoscopy may be needed. If a patient complains of bone pain or fractures, a radiographic skeletal survey or bone scan may be needed. On examination, it is important to look for lymphadenopathy and hepatosplenomegaly. If an abnormality is suspected, a liver ultrasound or CT scan should be ordered to investigate. If there are abnormal findings in any of the tests, a biopsy of the bone marrow should be considered. A complete blood count (CBC), serum tryptase level, liver function tests, and KIT gene analysis can be ordered. However, it is considered by some to be optional in the pediatric population. If there are any abnormalities, a bone marrow biopsy should be considered. Bone marrow involvement is not very common in children with cutaneous mastocytosis, unlike adults, and a bone marrow biopsy is not recommended. Tryptase levels may be more useful than histamine levels because histamine can be elevated in hypereosinophilic states. It is important to learn the criteria for systemic mastocytosis in case a patient progresses.[15]
The WHO criterion for the diagnosis of systemic mastocytosis states that you need 1 major and 1 minor criteria or 3 minor criteria.
Major Criteria
- Having multifocal, dense infiltrates of mast cells (aggregates ≥15 mast cells) in bone marrow or extracutaneous tissues.
Minor Criteria
- >25% of mast cells in bone marrow samples or extracutaneous tissues are spindle-shaped or otherwise atypical.
- Expression of CD25 and/or CD2 by extracutaneous mast cells (often determined by bone marrow flow cytometry)
- Presence of activating KIT codon 816 mutation in blood, bone marrow, or extracutaneous tissues.
- Serum total tryptase level >20 ng/ml (exception would be an associated clonal myeloid disorder, then this is not valid).
The criterion for diagnosing cutaneous mastocytosis is not well-defined. Cutaneous mastocytosis is usually diagnosed by visual evaluation of typical skin lesions, particularly in children. However, there is a stepwise approach to diagnose mastocytosis in the skin; it must have 1 major and 1 minor criterion.
Major Criteria
- Must have the typical skin lesions.
Minor Criteria
- Histology (monomorphic mast cell infiltrate with aggregates of >15 mast cells per cluster or scattered mast cells with >20 per high microscopic power field).
- Molecular criterion (detection of a KIT mutation at codon 816 in the affected skin).[13]
Treatment / Management
The management of cutaneous mastocytosis begins with practical measures, such as the use of lukewarm water for bathing, air conditioning during hot weather, and avoidance of triggers for mast cell degranulation. Treatment is symptomatic for patients with cutaneous mastocytosis.
Topical medications include calcineurin inhibitors and corticosteroids (a potent class 1 topical corticosteroid, with occlusion if required). Intralesional injections of small amounts of dilute corticosteroids may resolve skin lesions temporarily or indefinitely. The risk of skin atrophy and adrenocortical suppression resulting from the treatment is minimized by treating limited body areas during a single treatment session.
Systemic corticosteroids are useful only in specific situations, such as ascites, malabsorption, and severe skin disease. Case reports have described complete remission of bullous mastocytosis with oral corticosteroids,[16] but systemic therapy is often disappointing because the primary mode of action of corticosteroids is redistribution rather than the death of mast cells. Systemic therapies such as oral antihistamines (the mainstay of treatment), oral cromolyn sodium (explicitly used for GI symptoms), omalizumab, oral PUVA, narrowband ultraviolet B, and UVA1 can be used. A pre-measured epinephrine pen with an auto-injector should be given to all UP patients.
Cutaneous mastocytosis patients should avoid certain physical stimuli, including emotional stress, temperature extremes, physical exertion, bacterial toxins, envenomation by insects to which the patient is allergic, and rubbing, scratching, or traumatizing the lesions of cutaneous mastocytosis. Patients should also be educated to avoid possible triggers such as aspirin, nonsteroidal anti-inflammatory drugs, codeine, morphine, alcohol, thiamine, quinine, opiates, gallamine, decamethonium, procaine, radiographic dyes, dextran, polymyxin B, scopolamine, and D-tubocurarine.[17] Crawfish, lobster, spicy foods, hot beverages, and cheese should be avoided. However, the role of the previously mentioned foods is hypothetical at this point and needs further investigation.[18] One successful case report described the treatment of progressive c-KIT mutation cutaneous mastocytosis with imatinib, halting disease symptoms and progression and improving the length of the disease course.[19]
Differential Diagnosis
Bullous impetigo, secondary syphilis, carcinoid, leiomyoma, urticaria, juvenile xanthogranuloma, arthropod stings, and auto-immune bullous diseases are differential diagnoses. Increased mast cell numbers can be found in other inflammatory and neoplastic skin conditions, such as dermatofibromas, psoriasis, atopic dermatitis, and nevi. However, these disorders are also associated with additional characteristic histopathological changes in the skin that differ from those of mastocytosis.[20]
Prognosis
The prognosis for childhood mastocytosis is excellent, with 50% to 70% of patients remitting before adolescence. Cutaneous mastocytosis onset after the age of 10 years portends a poorer prognosis because the late-onset disease tends to be persistent, is associated more often with systemic disease, and carries a higher risk of malignant transformation.[21]
Complications
Complications may include the possible transformation into a hematologic malignancy (mast cell leukemia) and death secondary to extensive mast cell degranulation.
Deterrence and Patient Education
Deterrence involves educating patients about potential triggers like specific foods, medications, insect stings, or physical stimuli that can exacerbate their condition. By recognizing and avoiding these triggers, patients can mitigate symptom flare-ups and improve their overall quality of life. Simultaneously, patient education plays a vital role in helping individuals understand the nature of UP, emphasizing its typically benign course and the rarity of internal organ involvement. Patients are also educated about the importance of proper medication management, lifestyle modifications, and regular follow-up appointments. The significance of early intervention and regular medical check-ups to prevent complications associated with mast cell activation should be emphasized. Patients must be taught to recognize signs of systemic involvement and seek medical attention promptly. Through a combination of deterrence strategies and comprehensive patient education, healthcare professionals empower those with UP to take an active role in managing their condition effectively and enhancing their well-being.
Enhancing Healthcare Team Outcomes
Healthcare professionals, including physicians, advanced care practitioners, nurses, and pharmacists, need to develop the skills necessary for diagnosing and managing UP. This includes the ability to recognize the characteristic skin lesions and differentiate them from other skin conditions. Clinicians should also possess skills in conducting thorough patient assessments and utilizing appropriate diagnostic tests to confirm the diagnosis.
A strategic approach to managing patients with UP involves staying updated on the latest research and treatment options. Healthcare professionals should create individualized treatment plans, including medications, lifestyle modifications, and patient education to manage the condition effectively.
Effective interprofessional communication is crucial in managing UP. Physicians, nurses, pharmacists, and other healthcare professionals must collaborate to ensure a holistic approach to care. Sharing information, insights, and treatment plans promotes optimal patient outcomes and safety.
The interprofessional team should coordinate care for patients with UP. This involves ensuring that all team members are on the same page regarding the patient's diagnosis and treatment plan. Timely and accurate information exchange and clear roles within the care team are essential for enhancing patient-centered care and safety. This collaborative approach promotes comprehensive care and improves the overall performance of the healthcare team in managing patients affected by this unique skin disorder.