Continuing Education Activity
Vasculitis is a heterogeneous group of pathologies characterized by inflammation of vessels. They share common links of clinical, laboratory, and pathophysiologic features. The clinical and pathological features are variable and depend on the site and type of blood vessels affected. Vasculitis may occur as a primary process or may also be secondary to another underlying disease. Diseases in which vasculitis is a primary process are called primary systemic vasculitides. This activity reviews the evaluation and management of vasculitis and highlights the role of the interprofessional team in the recognition and management of this condition.
Objectives:
- Identify the etiologies of vasculitis medical conditions and emergencies.
- Review the appropriate process for examination and evaluation of vasculitis.
- Outline the treatment and management options available for vasculitis.
- Describe interprofessional team strategies for improving care coordination and communication to advance vasculitis and improve outcomes.
Introduction
Vasculitis is a heterogeneous group of pathologies characterized by inflammation of vessels. They share common links of clinical, laboratory, and pathophysiologic features. The clinical and pathological features are variable and depend on the site and type of blood vessels affected.[1] Research has identified more than 30 kinds of vasculitis.[2] Vasculitis may present as a primary process or secondary to another underlying disease. Diseases in which vasculitis is a primary process are called primary systemic vasculitides. Systemic vasculitides classify according to the 1990 American College of Rheumatology (ACR) criteria.[3] The International Chapel Hill Consensus Conference (CHCC) is the most commonly used nomenclature system for vasculitis.[2]
Etiology
The cause of various vasculitis is unknown — there is a recognition in the literature of several risk factors that increase the incidence and prevalence of vasculitis, including geography, age, ethnicity, gender, genetic, and environmental factors. Behcet disease is more common among inhabitants of countries that border the ancient Silk Route.[4] Takayasu disease is more prevalent in South Asian countries than elsewhere. Kawasaki disease is a disease of young children less than five years of age,[5] whereas giant cell arteritis (GCA) is a disease of the elderly. There is evidence that the incidence of GCA has increased over the past 50 years by a factor of 2 to 5.[6]
Takayasu disease is more common in females (9 to 1 ratio) than males, while Behcet disease tends towards higher severity in men, with a higher frequency of advanced ocular disease. GCA and granulomatosis with polyangiitis (GPA) occur predominantly in the White population.[7]
Studies have found the association of human leukocyte antigen (HLA)-B51 with Behcet disease, increasing the risk of disease susceptibility and disease severity.[4] Genetic studies have indicated roles for class II HLA alleles such as HLA-DRB1*0401 and HLA-DRB1*0101 in some studies for GCA.[8]
Several medications and certain infections have well-known associations with systemic vasculitis, e.g., hepatitis B with polyarteritis nodosa (PAN), hepatitis C with mixed cryoglobulinemia, silica dust with pauci-immune vasculitis.[9]
Epidemiology
The overall incidence per annum of primary systemic vasculitis is approximately 20 to 40/million, according to studies in Europe and the USA.[4] GCA is the most common among the primary vasculitides, with an annual incidence (in the population of individuals at least 50 years old) of around 240 per million.[10] Granulomatosis with polyangiitis (GPA), microscopic polyangiitis (MPA), and Churg-Strauss syndrome have been reported at an incidence anywhere from 1 to 10 per million.[4]
Pathophysiology
The endothelium is an active player in inflammatory processes and pathogenesis of vasculitis. Cytokine-mediated changes in the expression and function of adhesion molecules coupled with inappropriate activation of leukocytes and endothelial cells are the primary factors influencing the degree and location of vessel damage in vasculitis syndromes. Few mechanisms studied include pathogenic immune complex formation and deposition (mixed cryoglobulinemia, PAN), production of antineutrophilic cytoplasmic antibodies (in GPA, MPA, and Churg-Strauss syndrome), and pathogenic T lymphocyte responses and granuloma formation.[11]
Histopathology
Histopathology of biopsies in GCA show mononuclear cell infiltrate dominated by T lymphocytes and macrophages, with the inflammatory infiltrate penetrating all layers of the arterial wall. The infiltrates can be granulomatous with the accumulation of histiocytes and multinucleated giant cells (MGCs). Granuloma formation with multinucleated giant cells can be present in infiltrates & presence of MGCs correlates with increased risk for ischemic complications. Remodeling of the arterial wall leads to vascular complications associated with GCA, whereas fibrinoid necrosis is the hallmark of ANCA-associated vasculitis. Renal biopsies in the AAVs show focal, segmental lysis of glomerular tufts, disruption of the basement membrane, and accumulation of fibrinoid material. Churg-Strauss syndrome usually shows a granulomatous vasculitis of small arteries and veins associated with eosinophilic infiltration.[11]
History and Physical
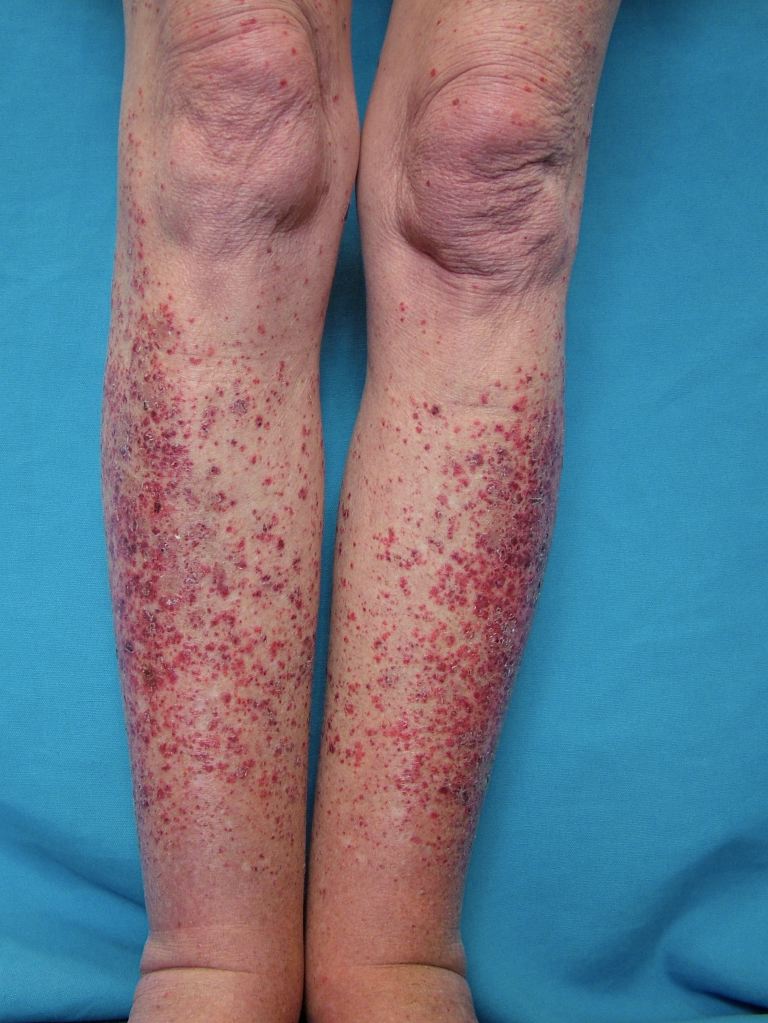
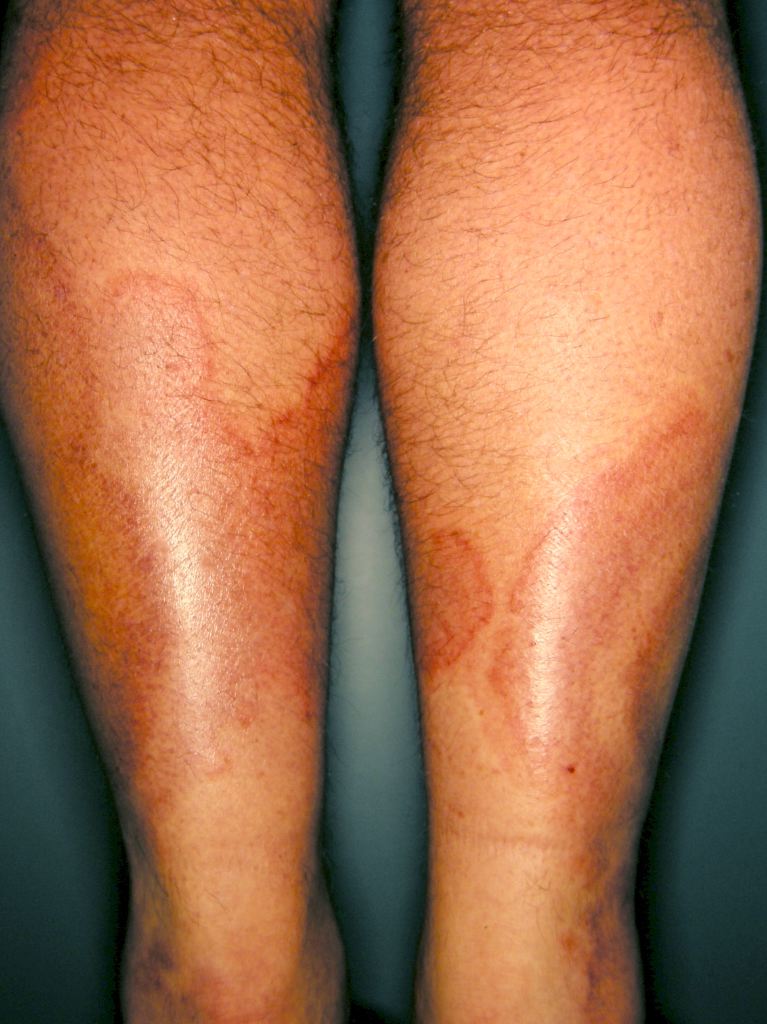
Systemic vasculitides are clinically heterogenous, so it is not possible to outline a single algorithm for evaluating patients suspected of vasculitis. A thorough medical history, a complete physical examination, and laboratory testing would be helpful as vasculitides are rare, but the potential exists for severe organ damage or death. Vasculitis should be a consideration in patients who present with systemic or constitutional symptoms in combination with evidence of single or multiorgan dysfunction. Suspicious findings in history include fevers, unexplained weight loss, nasal crusting, epistaxis, upper airway disease, ocular inflammation, acute foot drop or wrist drop, limb claudication, unexplained hemoptysis, hematuria, and a history of hepatitis. Suspicious findings on the physical exam include sensory or motor neuropathy, palpable purpura, nasal septal perforation, absent, diminished, or tender pulses, bruit, and blood pressure discrepancies.
Specific clinical abnormalities that, when present alone or in combination, should suggest a diagnosis of vasculitis. These include palpable purpura, pulmonary infiltrates and microscopic hematuria, chronic inflammatory sinusitis, mononeuritis multiplex, unexplained ischemic events, and glomerulonephritis with evidence of multisystem disease.
In this activity, the discussion will focus on GCA and ANCA-associated vasculitis (AAV).
Most common clinical manifestations for patients with GCA include fevers, weight loss, malaise, headaches, visual symptoms like amaurosis fugax, unilateral or bilateral vision loss, diplopia, jaw claudication, vertigo, and scalp tenderness. Atypical manifestations include throat pain, large artery disease, aortic dissection, neurologic signs like peripheral neuropathy, transient ischemic attack, stroke, myocardial infarction, delirium, etc.
AAV can affect any organ system, and symptoms usually depend on organ system involvement with different types of vasculitis. With GPA, patients can present with inflammation of the upper airway, epistaxis, sinus inflammation, and inflammation of the trachea or nasal cartilage, leading to saddle nose deformity. Eye and orbital inflammation are common in GCA and MPA as well. Pulmonary involvement in GCA, MPA, and CSS can present as causing hemoptysis, nodules, pleuritis, and alveolar hemorrhage. Neurologic manifestations in patients with AAV include peripheral neuropathy, sensorineural hearing loss, and CNS vasculitis. Glomerulonephritis (GN) can present early in the disease in about half of the patients with AAV.
Evaluation
When suspecting vasculitis workup should include a complete blood cell count (CBC) (usually associated with neutrophilia, anemia, and thrombocytosis), kidney function (may be abnormal in renal involvement from GPA, Churg-Strauss, etc.), liver function studies, erythrocyte sedimentation rate (ESR) and/or C-reactive protein (CRP), [elevation in acute phase reactants noticed in most vasculitis], serologies for viral hepatitis (hepatitis B in PAN), serum cryoglobulins, and urinalysis with urinary sediment. Additional testing should include anti-neutrophil cytoplasmic antibodies (ANCA), anti-nuclear antibodies (ANA), complement levels (low complement levels associated with mixed cryoglobulinemia), immunoelectrophoresis (monoclonal gammopathies can occur in Hepatitis C related vasculitis) and work up to rule out vasculitis mimics including infections.
A radiographic evaluation, such as a chest x-ray or high-resolution computed tomography (HR-CT), is indicated in patients with respiratory symptoms. Biopsy of the involved tissue is essential for the diagnosis of many vasculitides. A temporal artery biopsy should always be part of the workup in cases of suspected GCA. Skin biopsy of purpuric lesions and renal biopsies in patients with glomerulonephritis have high diagnostic yields. CT imaging of the sinuses may be necessary for patients with suspected GPA. Vascular imaging, including magnetic resonance imaging (MRI), MR angiograms, CT angiograms, vascular ultrasound, and positron emission tomography (PET), may be used to detect large artery lesions.
Treatment / Management
Treatment depends on the type of vasculitis. If an offending antigen is recognized, e.g., hepatitis infection, it should be treated appropriately along with the treatment regimen of vasculitis. Treatment regimens center upon the specific diagnosis and the severity or extent of the disease. The approach to treatment for any vasculitis generally includes three components: remission induction, remission maintenance, and monitoring.
Glucocorticoids are the first-line treatment for patients with vasculitis used with or without immunosuppressive agents. The type of vasculitis guides the choice of immunosuppressive agents. Methotrexate (MTX), azathioprine (AZA), mycophenolate (MMF), cyclophosphamide (CYC), rituximab (RTX), intravenous immunoglobulin, plasma exchange, etc., have all been used in various treatment regimens in different forms of vasculitis.[12] Once the condition is in remission, slow, downward glucocorticoid titration should commence to maintain control of disease activity and minimize the risks of drug toxicity. Patients and physicians should know the short-term and long-term toxic side effects of therapeutic agents for monitoring.
In this article, we will discuss the treatment and management of GCA, GPA, and MPA briefly.
Initial treatment for GCA includes steroids, starting at doses of 40 to 60 mg a day, as single or divided doses. Glucocorticoid treatment should be initiated without delay in patients with a strong suspicion for GCA. Intravenous pulse steroids are an option in patients with recent vision loss. After remission, steroids should be slowly tapered off. Methotrexate and azathioprine can serve as steroid-sparing agents. Tocilizumab has been approved recently for treatment of GCA
Patients with AAV have poor survival rates without treatment, with 81% mortality after one year of diagnosis. Current treatment regimens aim to induce remission and then maintain remission. Depending on disease severity, with organ- and life-threatening manifestations warranting the most aggressive therapy, treatments are tailored for various types of AAV. EULAR/ERA-EDTA published guidelines for the management of AAV in 2016, and patients with the non-organ threatening disease can start on a regimen of methotrexate or mycophenolate mofetil in combination with glucocorticoid. Patients with life-threatening disease should start on a regimen of cyclophosphamide or rituximab with glucocorticoids. Plasma exchange can be a consideration in patients with progressive renal failure or pulmonary hemorrhage. For maintenance therapy, regimens of azathioprine, mycophenolate mofetil, or rituximab with a tapering course of steroids have demonstrated efficacy.[13]
Differential Diagnosis
Vasculitis has several mimics, so careful evaluation of differentials is necessary when evaluating such patients. Conditions that can mimic vasculitis include infections such as infective endocarditis, histoplasmosis, and gonococcal infection. Since the treatment of vasculitis requires the use of immunosuppressive drugs, it is necessary to perform a full infection screen in all patients with suspected vasculitis. Coagulopathies like antiphospholipid syndrome & thrombotic thrombocytopenic purpura, neoplasms like atrial myoma, lymphoma, and several drug toxicities like cocaine and levamisole can present with symptoms similar to vasculitis.[14]
Prognosis
Long-term survival of patients with vasculitis highly depends on the diagnosis, response to therapy, and adverse effects of drugs, including the occurrence of infections. In a study assessing long-term survival in ANCA-associated vasculitis: the 1-, 2- and 5-year survival was 88%, 85%, and 78%, respectively. The mortality ratio was 2.6 compared with the general population.[15] Mortality reports derive from both active vasculitic disease and complications of therapy.
Complications
Complications of vasculitis are dependent on the type of vessel involved. Large vessel involvement with vasculitides like GCA, Takayasu, or Kawasaki can lead to complications like acute myocardial infarction and stroke, including ischemia of cranial arteries like the ophthalmic artery,[16] mesenteric ischemia, aortic syndromes (including dissection or intramural hematoma) or critical extremity ischemia.[17] Life-threatening complications of small-vessel vasculitis include alveolar hemorrhage, renal failure, and intestinal ischemia.[18] Aneurysm formation is a known complication of GCA, polyarteritis nodosa, and Behcet disease.[17] Deep venous thrombosis and pulmonary embolism are more frequent in AAV and Behcet disease than in other vasculitides.[4]
Deterrence and Patient Education
A patient should consult a physician if they notice any symptoms suspicious for vasculitis. If diagnosed with vasculitis, a patient will require a multi-disciplinary approach for management with the primary care physician and rheumatologist.
Enhancing Healthcare Team Outcomes
Vasculitis is a chronic systemic illness requiring an interprofessional team for its management. Shared decision-making and communication are crucial elements for a good outcome. Patients, rheumatologists, primary care physicians, nephrologists, ophthalmologists, pulmonologists, and other specialties, including pharmacists, would need to work closely, to achieve remission and improve the quality of care and quality of life for such patients.
Nursing will also play a key role in managing vasculitis cases, from administering medication to observing for disease or drug-related adverse effects and reporting these to the rest of the team. The pharmacist should verify all dosing and perform medication reconciliation to prevent drug-drug interactions, also alerting the team when these are present so a therapy change can occur proactively. The ordering/prescribing physician (MD, DO NP, PA) should work in concert with these other professions so that the interprofessional team model can drive optimal patient outcomes with minimal adverse events. [Level 5]