Introduction
The development of the eye is a complex multifactorial symphony of signaling molecules, receptors, and molecular gradients organized in a melody of the time and space during embryogenesis. Understanding this process within the eye allows us to have a better understanding of the basis of congenital eye diseases. The optic cup is particularly important to the proper formation of the eye, as it forms all major structures of the globe, except for the lens. Congenital glaucoma, retinal detachment, and coloboma are phenomena related to defects in the optic cup or its associated structures, and all of these can have serious consequences for the sight of the patient.
Development
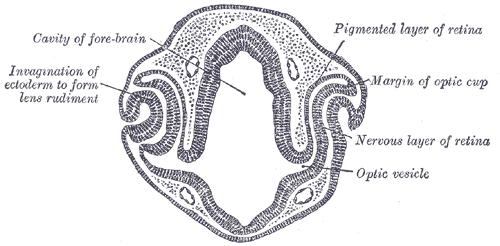
The development of the primitive eye begins at approximately week 3 of gestation with the burgeoning of the optic sulcus from the periocular mesenchyme. The optic sulci are bilateral invaginations on both sides of the anterior neural tube at the diencephalon. As the sulci deepen, they become optic pits and eventually transform into the optic vesicles as they extend toward surface ectoderm. Both surface ectoderm and the optic vesicles secrete/excrete extracellular matrix and adhere to each other. As the optic vesicles extend outward, the proximal attachment to the forebrain becomes the optic stalk. The optic vesicles aid in the migration of neural crest cells, which contribute to future development. At approximately 5 weeks, a portion of the optic vesicle will undergo auto-invagination to produce a “cup” with two cell layers. This invagination is asymmetric, and a groove forms in the inferior border. This structure is called the optic fissure, and early vasculature and periocular mesenchyme are introduced to the eye via this structure. The optic fissure closes by the end of week 6.[1][2][3][4]
The two layers forming the optic cup is a continuous tissue that gives bends at a hinge point known as the optic cup lip, and that marks the border of the outer pigmented layer and inner neural layer.[5] The anterior portion of the optic cup gives rise to the pars caeca retinae.[6] This structure, in turn, will go on to form the ciliary body, iris, and pupillary muscles.[7] The posterior portion of the optic cup forms the retina.[8] The outer layer of the optic cup forms the retinal pigmented epithelium. The inner layer of the optic cup eventually forms the outer nuclear layer (which contains rods and cones), the inner nuclear layer (which contains bipolar cells), and the ganglion layer (which contains ganglion cells). During week 7, axons begin to grow into the optic stalk, and development of the optic nerve is completed by week 8.
Cellular
Neuroectoderm cells comprise the optic cup.[9] While structures that are direct derivatives of the optic cup are in the above section, it is important to note the embryologic origin of other ocular structures. Surface ectoderm is responsible for the development of the lens, conjunctiva, lids, and corneal epithelium. The optic cup lip becomes the adult iris edge. Neural crest cells give rise to the corneal endothelium, trabecular meshwork (TM), scleral fibroblasts, connective fascia of extraocular muscles, and optic nerve meninges.
Molecular Level
Multiple genes have been shown to interact during the development of the primitive eye, as is the case with the rest of the developing embryo. In the eye, the PAX6 gene is the primary control gene, with the WNT gene and FGF gene playing a supplementary role during the development of the optic vesicles. Another prominent gene in embryonic development of the eye is the Sonic hedgehog (Shh) gene, which is responsible for the suppression of PAX6. If Shh becomes inhibited, then PAX6 deregulation results in cyclopia. Conversely, overexpression of Shh has been shown to result in a loss of eye structures. Retinoic acid also plays an important role in the development of the eye, mainly through its use as a paracrine inhibitor of the mesenchyme around the optic cup. Lack of retinoic acid can thus lead to defects in the anterior segment and results in blindness.
However, the interplay between genes and molecular signaling factors are so complex that it does not fit in the scope of this article. Some of the other genes involved in development include but are not limited to PITX2, PITX3, FOXC1, FOXE3, LMX1B, GPR48, TFAP2A, and TFAP2B.
Function
The optic cup gives rise to the entire globe, along with its associated inner structures except for the lens, which includes the ten layers of the retina that contains rods, cones, bipolar cells, amacrine cells, horizontal cells, and the complex interplay between these cells that help process photons into three-dimensional vision.
Clinical Significance
Because the optic cup gives rise to so many ocular structures, it has broad clinical significance. One classic example of a condition that can result from defective optic cup embryogenesis is coloboma. A coloboma is a defect with absent tissue in part of an ocular structure. Colobomas are thought to be caused by failed closure of the optic fissure.[4] Coloboma can occur in most eye structures and are most commonly associated with defects in the iris, cornea, retina, optic nerve, or choroid.[9] Coloboma is associated with microphthalmia[9], and may cause blindness; it accounts for up to 2% of blindness in adults,[10] and up to 11% of blindness in children.[9] The co-occurrence of microphthalmia is unclear but may be due to globe ectasia from the coloboma.
Axenfeld-Rieger syndrome (ARS) is a rare genetic condition that may be due to defects affecting the optic cup. ARS is an autosomal dominant syndrome that has multiple effects on the anterior segment and may lead to glaucoma.[11] Many abnormalities may be present in ARS; findings may occur in conjunction with one another or an isolated manner.[12] Findings are generally broken up into three categories: those affecting the iris, cornea, and chamber angle. Iris defects include hypoplasia, corectopia, and polycoria.[12] Posterior embryotoxon is the most common corneal sign of ARS but is not seen in all patients with the syndrome; however, the presence of posterior embryotoxon warrants investigation for ARS. In the chamber angle, iris strands may be seen bridging from the iridocorneal angle to the TM. A major mutation associated with ARS is in the transcription factor PITX2. A specific g-protein-coupled receptor (GPR48) that is found extensively in the optic cup is thought to regulate PITX2.[13] Thus, mutations in GPR48 may result in ARS and cause glaucoma in these patient populations.
Defects in the development of the optic cup also contribute to numerous rare ocular diseases.[13] Because there is a complex interplay between the various embryologic tissue types that form ocular structures (of which the optic cup is neuroectoderm), conditions of structures that are defects in the neural crest or surface ectoderm in nature may be influenced by abnormal neuroectodermal signaling. However, an exploration of all rare diseases influenced by neuroectodermal signaling without express defects in neuroectodermal structures is beyond the scope of this activity.
Morning glory syndrome is another rare anomaly that may be due to optic cup dysgenesis.[14] This anomaly may present with retinal detachment, a glial tuft on the optic nerve, and microphthalmos.[15]
Retinoic acid is essential to proper optic cup development. Thus deficiency or mutations in retinoic acid signaling can lead to anomalous development; this can range from relatively mild disorders, such as Fundus albipunctatus, to very severe disorders such as Matthew-Wood syndrome or Leber congenital amaurosis.[16]
In summary, the optic cup is an embryologic structure that gives rise to many structures in the globe. Also, signaling from the optic cup leads to the development of additional intra-ocular and extra-ocular structures. Dysgenesis can result in a wide array of disorders, from retinal detachment to glaucoma to anterior segment dysgenesis.