Introduction
Desquamative interstitial pneumonia (DIP) is a relatively uncommon form of idiopathic interstitial pneumonia (IIP), classified according to international multidisciplinary consensus guidelines.[1] The term "desquamative interstitial pneumonia or DIP" originated from studies in the early 1960s that described alveolar infiltrates resulting from alveolar epithelial cell desquamation.[2] However, later research revealed that these infiltrates primarily consist of alveolar filling with pigmented macrophages.
Etiology
Register For Free And Read The Full Article
 Search engine and full access to all medical articles
Search engine and full access to all medical articles- 10 free questions in your specialty
- Free CME/CE Activities
- Free daily question in your email
- Save favorite articles to your dashboard
- Emails offering discounts
Learn more about a Subscription to StatPearls Point-of-Care
Etiology
The exact etiology of DIP remains unknown. While smoking is a known risk factor, its relationship with DIP is less pronounced compared to other smoking-related lung disorders, such as chronic obstructive pulmonary disease. A surgical lung biopsy is required for diagnosing DIP, as clinical and radiological findings alone are insufficient.
In 1965, Liebow et al first reported this disease and, based on their interpretation of large cells within alveolar spaces as desquamated epithelial cells from the lining walls, named it DIP.[3] However, this name has since been proven to be a misnomer, as the cells were identified as macrophages within the air spaces. The original authors did not mention the distinctive association with smoking, which was first highlighted by Carrington et al in 1978.[4][5]
DIP has historically been linked to tobacco smoking; however, later reports revealed that up to 40% of cases occur in lifelong nonsmokers, suggesting other potential causes.[6] DIP has also been associated with environmental exposures and autoimmune conditions,[7] including:
- Inhalation of or exposure to dust or fumes.[8]
- Inhalation of marijuana.[6][9]
- Exposure to metals, such as copper and beryllium, affects workers such as tool grinders, arc polishers, aluminum arc welders, machinists, and tire manufacturing workers.[8][10][11][12]
- Occupational exposure to fire extinguisher powder.
- Nylon filament exposure in textile workers.[13]
- Use of medications such as sirolimus, nitrofurantoin, tocainide, and sulfasalazine.[14]
- Autoimmune diseases such as scleroderma and rheumatoid arthritis.
- Recurrent episodes following lung transplant.
DIP has been associated with infectious etiologies such as Aspergillus, hepatitis C, and cytomegalovirus, as well as metabolic disorders such as Gaucher disease.[15][16] In addition, recent evidence suggests that new molecular defects, such as granulocyte-macrophage–colony-stimulating factor (GM-CSF), may contribute to alveolar macrophage accumulation, particularly following tobacco smoking exposure. This indicates that GM-CSF could have a role in the pathogenesis of DIP.[17] Although adult-onset DIP is not associated with any genetic defects, pediatric DIP is a distinct clinical entity linked to genetic mutations in surfactant proteins B and C, leading to dysfunction.[18]
Epidemiology
The prevalence of DIP is unknown.[19] Due to the rarity of the condition, most epidemiological studies include data from idiopathic pulmonary fibrosis (IPF), with an estimated prevalence of 3 to 6 per 100,000 cases.[20][21] The most common age of onset is between 40 and 60, with a reported male-to-female ratio of 2:1. The prevalence of smoking among patients ranges from 60% to 87%.[10][22][23] Craig et al and Tubbs et al independently reported about a 40% prevalence of nonsmokers in their cohorts.[10][23]
Pathophysiology
Over the last few decades, the identification of respiratory bronchiolitis–interstitial lung disease (RB-ILD) has led to increased examination of DIP pathophysiology, with scientific communities now considering the 2 disorders as part of a continuous spectrum.[24] The histological findings related to RB-ILD indicate a heightened inflammatory response, characterized by a significant increase in macrophages around and within the respiratory bronchioles, accompanied by mild interstitial thickening.
On the other hand, the histological characteristics of DIP are believed to involve uniform, diffuse airspace occupancy by macrophages, along with mild interstitial thickening and a mild, chronic inflammatory infiltrate (see Image. Pathological Features of Desquamative Interstitial Pneumonia).
Histopathology
In contrast to respiratory bronchiolitis or RB-ILD, macrophage accumulation in DIP is extensive, diffuse, and uniform within the alveolar spaces. This extensive infiltration of macrophages results in eosinophilic staining due to the abundant eosinophilic cytoplasm of the macrophages. Alveolar macrophages often contain a distinct light brown pigmentation that does not stain for hemosiderin, commonly referred to as "smoker's pigment." These macrophages are known as "smoker's macrophages." In marijuana smokers with DIP, the macrophages typically contain smaller, golden-brown particulate material.[9]
In addition, the alveolar septa are infiltrated by lymphocytic conglomerates, lymphoid follicles, and even eosinophils. Radiologically, cystic spaces may appear in areas of ground-glass opacities. These small cystic air spaces observed in chest imaging in DIP have been shown to correlate histologically with dilated alveolar ducts and bronchiectasis.[25]
Overall, the alveolar architecture is well preserved, but focal interstitial thickening and inflammatory changes are common. In the later stages of the disease, the production of transforming growth factor beta-1 (TGFβ1) leads to fibrosis around the alveolar interstitium. However, unlike usual interstitial pneumonia (UIP), fibroblastic foci are rarely observed. Multinucleated giant cells are frequently reported.
History and Physical
Clinical and histopathological features often overlap in smoking-related interstitial pneumonia. As a result, a consensus based on clinical, radiological, and pathological findings during the period of longitudinal follow-up is essential to accurately characterize the specific form of ILD and guide appropriate treatment.
Individuals with a history of relevant exposure to cigarette use, inhalational drugs of abuse, and occupational risk factors commonly present with a dry cough and exercise-induced shortness of breath. Although childhood disease has been rarely reported, as mentioned above, the most common age of presentation is between 40 and 60, predominantly in males.[4] Less than 10% of patients are asymptomatic at diagnosis. Symptoms are nonspecific and include dyspnea on exertion (90%), persistent cough (70%), with or without sputum production (about 40%). Hemoptysis is very rare.[6][26] Clinical examination often reveals clubbing in 50% of patients, along with coarse rales in bilateral bases.
Evaluation
The initial evaluation of patients suspected of DIP involves removing any iatrogenic or environmental exposures. If there is no clinical recovery, the evaluation includes basic serology tests (antinuclear antibodies, C-reactive protein, and rheumatoid factor), which are typically negative in DIP, as well as a peripheral blood count with differential, which may show an increase in neutrophil and eosinophil count (see Image. Approach to the Evaluation of Suspected Desquamative Interstitial Pneumonia).
Pulmonary function tests (PFTs) typically reveal a restrictive defect with impaired diffusion capacity, differing from the obstructive pattern seen in respiratory bronchiolitis.
A chest x-ray may reveal a reticulonodular infiltrate in the lower lung zones, though it is often subtle and nonspecific. In 3% to 22% of biopsy-proven cases, the chest x-ray may appear normal.
High-resolution computed tomography (HRCT) scan of the chest is the imaging modality of choice for evaluating DIP. HRCT typically shows diffuse, bilateral, lower-lobe predominant ground glass opacities in a homogeneous distribution, without honeycombing—distinguishing it from UIP or idiopathic pulmonary fibrosis (IPF). In progressive cases, cysts and traction bronchiectasis may develop, although they are not exclusively subpleural. Fibrosis is less common in DIP compared to nonspecific interstitial pneumonia (NSIP) or UIP. However, advanced DIP can be indistinguishable from NSIP due to a temporally uniform pattern of fibrosis that develops beyond the initial accumulation of alveolar macrophages. Sugiyama and colleagues reported that late-stage association occurred in 30% of cases.[27]
Unlike other inhalational lung diseases, ground glass infiltrates in DIP are more predominant in the lower lung zones. Interestingly, a study by Cartier and colleagues found that radiologists' diagnosis of DIP based on HRCT was histologically confirmed in only 59% of cases, highlighting the importance of a clinical, radiological, and histological consensus.[28]
In patients with a pattern not consistent with UIP on HRCT, cellular analysis of bronchoalveolar lavage (BAL) fluid can help differentiate DIP from eosinophilic pneumonia and sarcoidosis. A markedly elevated eosinophil count is more indicative of eosinophilic pneumonia, while a significantly elevated proportion of lymphocytes and a high CD4-to-CD8 ratio are more suggestive of sarcoidosis.[29]
Transbronchial biopsy (TBBX) may not provide representative tissue sections and is often associated with findings consistent with organizing pneumonia in early stages or fibrotic changes in later stages. If TBBX is nondiagnostic, a surgical lung biopsy, with an adequate tissue sample to assess the predominant histological features, is considered the gold standard for diagnosing DIP.
In recent years, transbronchial cryobiopsy has been explored as an alternative diagnostic approach, offering an increased sample size through noninvasive means.[30] Studies have repeatedly shown considerable overlap between RB-ILD and DIP, with features of both potentially present in different sections of the biopsy.[11][31][32] However, radiological manifestations across the entire lung field and PFT abnormalities differ significantly between the 2 conditions.
Treatment / Management
The most crucial intervention following diagnosis is smoking cessation. If the condition is linked to specific occupational exposures, avoiding further exposure is essential to prevent disease progression. In addition to eliminating exposure and risk factors, preventive measures commonly recommended for other chronic ILDs include vaccinations against pneumococcal pneumonia, respiratory syncytial virus, coronavirus, and influenza. Other supportive measures include oxygen therapy to maintain oxygen saturation levels between 90% and 94%.
Systemic corticosteroid therapy over several months has been reported as the most effective pharmacological intervention for DIP. With corticosteroids and immunosuppressive therapy (azathioprine being the most commonly used agent), most patients remain stable or show improvement, with some even experiencing complete recovery.[33][34] However, studies by both Aubry et al and Kawabata et al have shown that approximately 25% of patients may continue to progress despite corticosteroid therapy, leading to pulmonary fibrosis.[35][36] In patients refractory to steroids, there have been reports of successful treatment with clarithromycin.[37](B2)
Isolated case reports describe the use of azathioprine and cyclophosphamide to treat acute respiratory failure due to DIP. In cases of progressive and severe lung disease causing hypoxemia, pulmonary hypertension, and significant reductions in total lung capacity or diffusing capacity for carbon monoxide, lung transplantation evaluation is indicated. However, reports indicate that DIP can recur after lung transplantation.[6][38][39]
Differential Diagnosis
The differential diagnoses include other smoking-related IIPs, such as RB-ILD, as well as other forms of IIP and lung diseases, including:
- Idiopathic pulmonary fibrosis
- Idiopathic nonspecific interstitial pneumonia
- Cryptogenic organizing pneumonia
- Acute interstitial pneumonia (formerly Hamman-Rich syndrome)
- Idiopathic lymphoid interstitial pneumonia
- Idiopathic pleuroparenchymal fibroelastosis
- Amiodarone toxicity
- Eosinophilic pneumonia
- Sarcoidosis
Prognosis
The overall survival rate for DIP is 70% to 88% after 10 years.[4][24] Mortality in DIP ranges anywhere between 6% and 28%.[4][33] Although spontaneous improvement has been reported, two-thirds of DIP patients experience disease progression if left untreated. In a study by Aubry et al, 25% of patients continued to show progressive disease despite corticosteroid therapy.[36]
Few longitudinal studies have characterized the rate of disease progression in DIP. In one such study, Remy-Jardin et al reported an increase in ground-glass changes, emphysema, and centrilobular nodules from 28% to 42%, 26% to 40%, and 33% to 35%, respectively, in current smokers during a mean follow-up period of 5.5 years.[40] In a radiological follow-up of DIP in a small group of 14 patients, cysts increased, and new honeycombing appeared in 35% of cases, including patients on systemic corticosteroid therapy.[35] Survival rates at 10 years are reported to be approximately 70%.[41]
Continuation of smoking is clearly associated with a poorer outcome.[24][42] Children with DIP tend to have a poorer prognosis, particularly for those with ABCA3 mutations.[43][44][45]
Complications
Acute interstitial pneumonitis and diffuse alveolar hemorrhage have been reported as acute complications of the disease.[46] Notably, there have been reported cases of acute exacerbations after diagnostic video-assisted thoracoscopic surgery.[47][48]
A debate exists regarding the progression of DIP into the domain of fibrotic NSIP in advanced stages, particularly concerning histopathological features. However, the prognosis in this group is generally better than that seen in either NSIP or UIP.
Deterrence and Patient Education
Patient education plays a crucial role in managing DIP, particularly since smoking is a significant risk factor for the disease. Patients should be educated on the strong association between tobacco use and DIP, emphasizing smoking cessation as the most effective preventive measure and a critical component of treatment. Healthcare providers should provide resources for smoking cessation, including counseling and pharmacologic aids, to support patients in quitting.
Additionally, patients should be informed about the importance of avoiding environmental exposures that may contribute to lung inflammation, such as occupational dust, fumes, and air pollutants. Regular follow-ups, pulmonary function testing, and imaging can help monitor disease progression, reinforcing the need for adherence to treatment plans. Educating patients about symptoms such as progressive dyspnea and cough ensures early recognition and prompt intervention, potentially improving long-term outcomes.
Pearls and Other Issues
Key facts to keep in mind regarding "desquamative interstitial pneumonia" include:
- Respiratory bronchiolitis or RB-ILD and DIP are well-characterized forms of smoking-related interstitial lung disease.
- DIP is distinguished from RB-ILD by its uniform, homogeneous pattern of pneumonitis across lung fields, whereas RB-ILD primarily exhibits a bronchiolocentric distribution.
- Histologically, DIP is characterized by the intra-alveolar accumulation of "smoker's macrophages" with eosinophilic cytoplasm and characteristic brown "smoker's pigment."
- Unlike UIP, the interstitial involvement in DIP is temporally homogeneous and not subpleural, with fibrosis being a relatively minor feature. Honeycombing is rarely seen on imaging.
- Although a surgical lung biopsy remains the gold standard for diagnosis, transbronchial cryobiopsy is being explored as a potential alternative.
- Immediate cessation of smoking or removal from occupational exposures is crucial to halting disease progression. Corticosteroids, with or without additional immunosuppressants, form the cornerstone of pharmacotherapy, with isolated reports suggesting using clarithromycin in refractory cases.
- Despite treatment, 25% of patients progress to fibrotic disease, often making it indistinguishable from NSIP.
- Continued smoking in adults, as well as childhood disease, are known to be poor prognostic indicators.
Enhancing Healthcare Team Outcomes
Enhancing healthcare team outcomes for desquamative interstitial pneumonia (DIP) requires a multidisciplinary approach focused on early diagnosis, effective management, and patient-centered care. The incidence and prevalence of DIP are so rare that large population-based randomized controlled trials are not available to guide management and outcomes. However, the association with cigarette and marijuana smoking, as well as certain occupational exposures, has been unequivocally established. Once diagnosed, discontinuation of exposure becomes the most important intervention, as emphasized by the 2013 American Thoracic Society and European Respiratory Society statement. Since smoking is a major risk factor, integrating smoking cessation programs within clinical practice, along with patient education, counseling, and pharmacologic support, is critical for improving outcomes.[49]
Given the considerable overlap between smoking-related interstitial lung diseases such as RB-ILD, DIP, and NSIP, a combined clinical, radiological, and histopathological approach to diagnosis is essential. Mixed histopathological features are frequently reported, necessitating longitudinal follow-up to distinguish between these entities. Prognosis varies among RB-ILD, DIP, and NSIP, with increasing severity in that order. Mortality is rarely reported for RB-ILD, with disease regression observed over 30 years following smoking cessation. In contrast, DIP has a reported mortality rate of 6% to 28%, and disease progression can occur even with treatment and smoking cessation.[50]
Media
(Click Image to Enlarge)
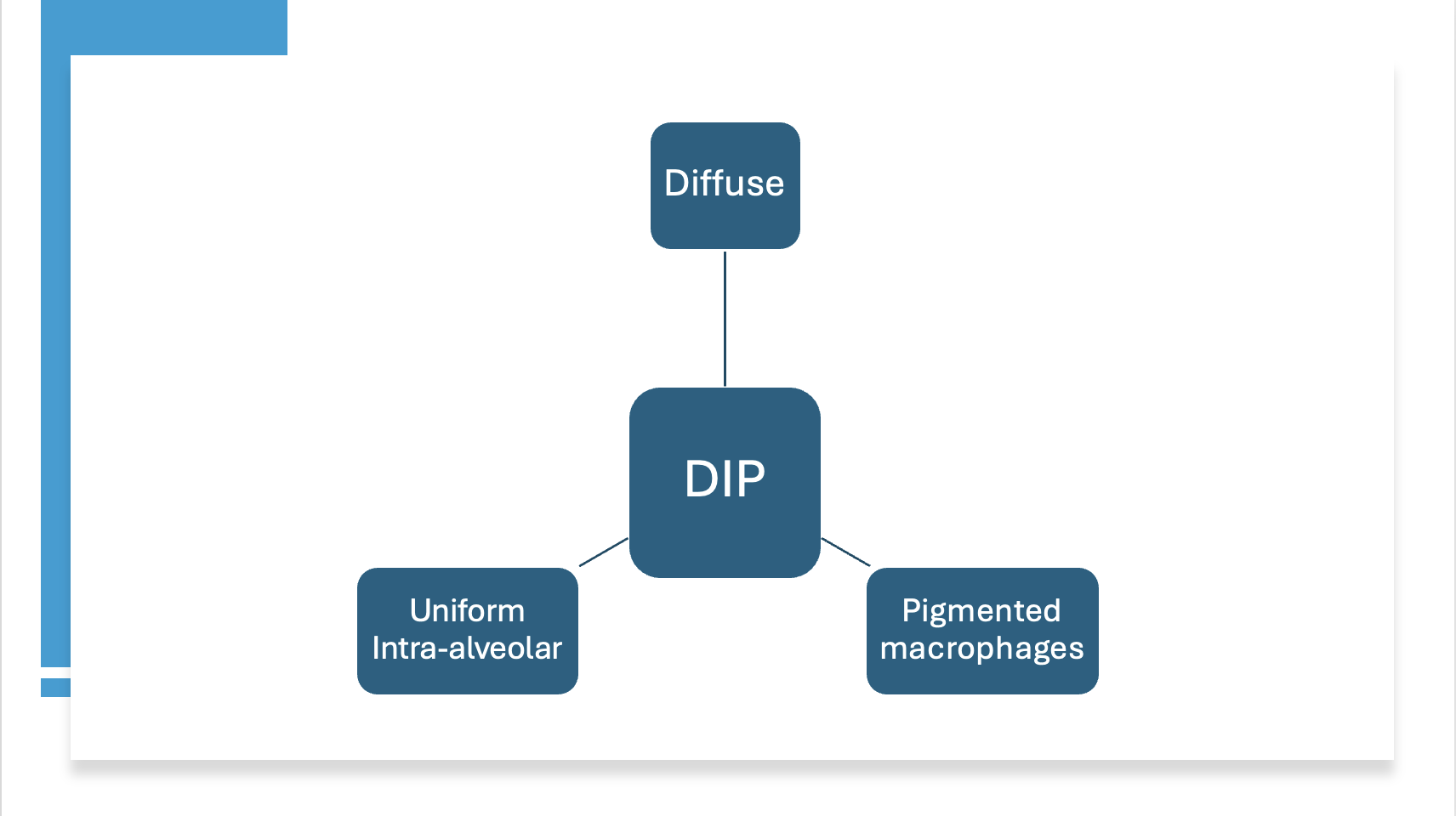
Pathological Features of Desquamative Interstitial Pneumonia. Abbreviation: DIP, desquamative interstitial pneumonia.
Contributed by A Sankari, MD
(Click Image to Enlarge)
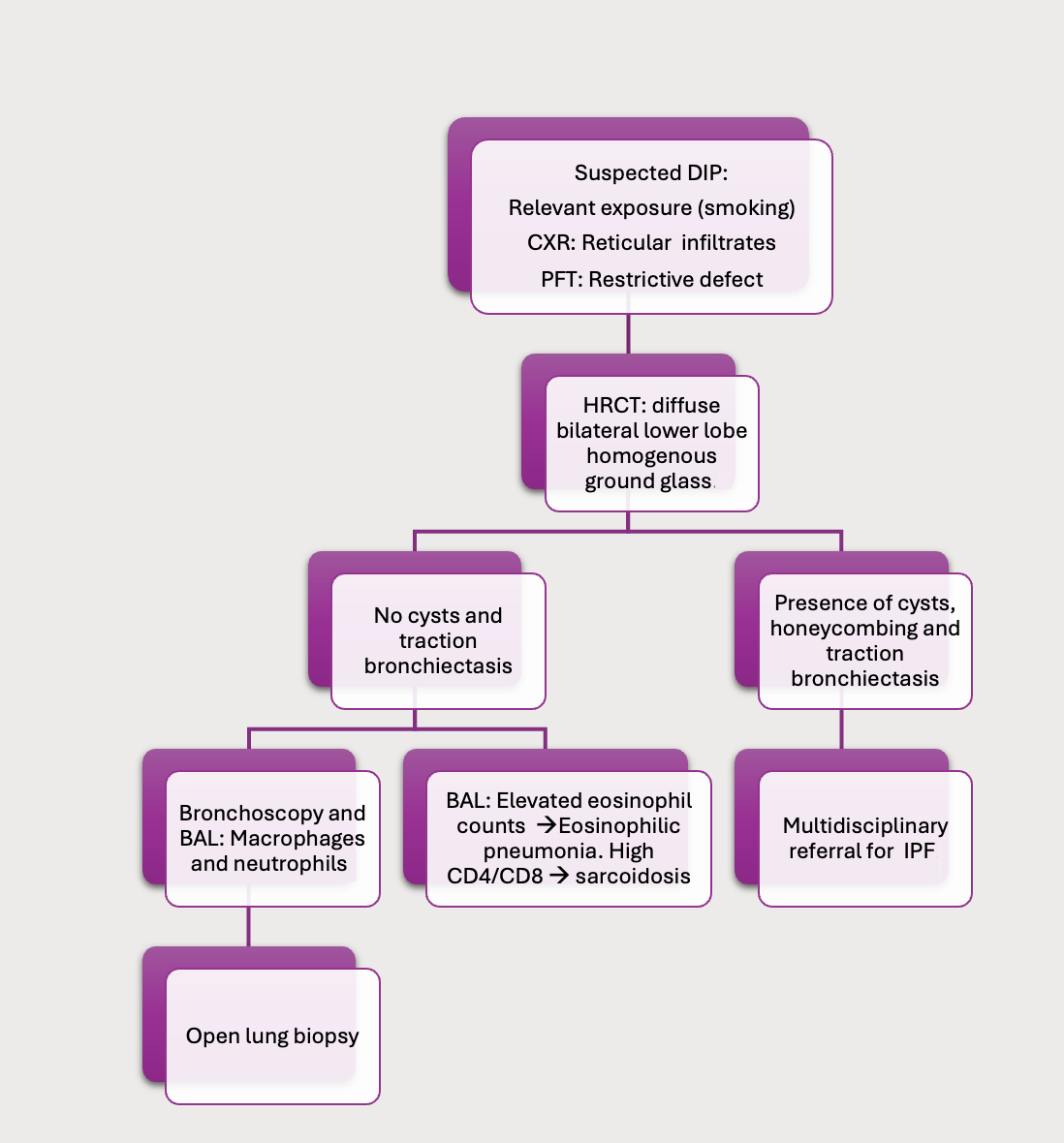
Approach to the Evaluation of Suspected Desquamative Interstitial Pneumonia. Abbreviations: BAL, bronchoalveolar lavage; CXR, chest x-ray; DIP, desquamative interstitial pneumonia; HRCT, high-resolution computed tomography; IPF, idiopathic pulmonary fibrosis; PFT, pulmonary function test.
Contributed by A Sankari, MD
References
Travis WD, Costabel U, Hansell DM, King TE Jr, Lynch DA, Nicholson AG, Ryerson CJ, Ryu JH, Selman M, Wells AU, Behr J, Bouros D, Brown KK, Colby TV, Collard HR, Cordeiro CR, Cottin V, Crestani B, Drent M, Dudden RF, Egan J, Flaherty K, Hogaboam C, Inoue Y, Johkoh T, Kim DS, Kitaichi M, Loyd J, Martinez FJ, Myers J, Protzko S, Raghu G, Richeldi L, Sverzellati N, Swigris J, Valeyre D, ATS/ERS Committee on Idiopathic Interstitial Pneumonias. An official American Thoracic Society/European Respiratory Society statement: Update of the international multidisciplinary classification of the idiopathic interstitial pneumonias. American journal of respiratory and critical care medicine. 2013 Sep 15:188(6):733-48. doi: 10.1164/rccm.201308-1483ST. Epub [PubMed PMID: 24032382]
American Thoracic Society, European Respiratory Society. American Thoracic Society/European Respiratory Society International Multidisciplinary Consensus Classification of the Idiopathic Interstitial Pneumonias. This joint statement of the American Thoracic Society (ATS), and the European Respiratory Society (ERS) was adopted by the ATS board of directors, June 2001 and by the ERS Executive Committee, June 2001. American journal of respiratory and critical care medicine. 2002 Jan 15:165(2):277-304 [PubMed PMID: 11790668]
Level 3 (low-level) evidenceLIEBOW AA, STEER A, BILLINGSLEY JG. DESQUAMATIVE INTERSTITIAL PNEUMONIA. The American journal of medicine. 1965 Sep:39():369-404 [PubMed PMID: 14338290]
Carrington CB, Gaensler EA, Coutu RE, FitzGerald MX, Gupta RG. Natural history and treated course of usual and desquamative interstitial pneumonia. The New England journal of medicine. 1978 Apr 13:298(15):801-9 [PubMed PMID: 634315]
Carrington CB, Gaensler EA, Coutu RE, Fitzgerald MX, Gupta RG. Usual and desquamative interstitial pneumonia. Chest. 1976 Feb:69(2 Suppl):261-3 [PubMed PMID: 1248298]
Level 2 (mid-level) evidenceGodbert B, Wissler MP, Vignaud JM. Desquamative interstitial pneumonia: an analytic review with an emphasis on aetiology. European respiratory review : an official journal of the European Respiratory Society. 2013 Jun 1:22(128):117-23. doi: 10.1183/09059180.00005812. Epub [PubMed PMID: 23728865]
Ishii H, Iwata A, Sakamoto N, Mizunoe S, Mukae H, Kadota J. Desquamative interstitial pneumonia (DIP) in a patient with rheumatoid arthritis: is DIP associated with autoimmune disorders? Internal medicine (Tokyo, Japan). 2009:48(10):827-30 [PubMed PMID: 19443979]
Abraham JL, Hertzberg MA. Inorganic particulates associated with desquamative interstitial pneumonia. Chest. 1981 Jul:80(1 Suppl):67-70 [PubMed PMID: 7249745]
Gill A. Bong lung: regular smokers of cannabis show relatively distinctive histologic changes that predispose to pneumothorax. The American journal of surgical pathology. 2005 Jul:29(7):980-2 [PubMed PMID: 15958866]
Level 3 (low-level) evidenceTubbs RR, Benjamin SP, Reich NE, McCormack LJ, Van Ordstrand HS. Desquamative interstitial pneumonitis. Cellular phase of fibrosing alveolitis. Chest. 1977 Aug:72(2):159-65 [PubMed PMID: 884976]
Moon J, du Bois RM, Colby TV, Hansell DM, Nicholson AG. Clinical significance of respiratory bronchiolitis on open lung biopsy and its relationship to smoking related interstitial lung disease. Thorax. 1999 Nov:54(11):1009-14 [PubMed PMID: 10525560]
Level 2 (mid-level) evidenceLougheed MD, Roos JO, Waddell WR, Munt PW. Desquamative interstitial pneumonitis and diffuse alveolar damage in textile workers. Potential role of mycotoxins. Chest. 1995 Nov:108(5):1196-200 [PubMed PMID: 7587416]
Dvorácková I, Píchová V. Pulmonary interstitial fibrosis with evidence of aflatoxin B1 in lung tissue. Journal of toxicology and environmental health. 1986:18(1):153-7 [PubMed PMID: 3084802]
Hamadeh MA, Atkinson J, Smith LJ. Sulfasalazine-induced pulmonary disease. Chest. 1992 Apr:101(4):1033-7 [PubMed PMID: 1348220]
Iskandar SB, McKinney LA, Shah L, Roy TM, Byrd RP Jr. Desquamative interstitial pneumonia and hepatitis C virus infection: a rare association. Southern medical journal. 2004 Sep:97(9):890-3 [PubMed PMID: 15455981]
Level 3 (low-level) evidenceSung SA, Ko GJ, Kim JY, Kim MG, Lee JE, Kim GI, Jo SK, Cho WY, Kim HK. Desquamative interstitial pneumonia associated with concurrent cytomegalovirus and Aspergillus pneumonia in a renal transplant recipient. Nephrology, dialysis, transplantation : official publication of the European Dialysis and Transplant Association - European Renal Association. 2005 Mar:20(3):635-8 [PubMed PMID: 15735246]
Level 3 (low-level) evidenceSuzuki T, McCarthy C, Carey BC, Borchers M, Beck D, Wikenheiser-Brokamp KA, Black D, Chalk C, Trapnell BC. Increased Pulmonary GM-CSF Causes Alveolar Macrophage Accumulation. Mechanistic Implications for Desquamative Interstitial Pneumonitis. American journal of respiratory cell and molecular biology. 2020 Jan:62(1):87-94. doi: 10.1165/rcmb.2018-0294OC. Epub [PubMed PMID: 31310562]
Avital A, Hevroni A, Godfrey S, Cohen S, Maayan C, Nusair S, Nogee LM, Springer C. Natural history of five children with surfactant protein C mutations and interstitial lung disease. Pediatric pulmonology. 2014 Nov:49(11):1097-105. doi: 10.1002/ppul.22971. Epub 2013 Dec 17 [PubMed PMID: 24347114]
Diken ÖE, Şengül A, Beyan AC, Ayten Ö, Mutlu LC, Okutan O. Desquamative interstitial pneumonia: Risk factors, laboratory and bronchoalveolar lavage findings, radiological and histopathological examination, clinical features, treatment and prognosis. Experimental and therapeutic medicine. 2019 Jan:17(1):587-595. doi: 10.3892/etm.2018.7030. Epub 2018 Nov 29 [PubMed PMID: 30651839]
Scott J, Johnston I, Britton J. What causes cryptogenic fibrosing alveolitis? A case-control study of environmental exposure to dust. BMJ (Clinical research ed.). 1990 Nov 3:301(6759):1015-7 [PubMed PMID: 2249047]
Level 2 (mid-level) evidenceDemedts M, Wells AU, Antó JM, Costabel U, Hubbard R, Cullinan P, Slabbynck H, Rizzato G, Poletti V, Verbeken EK, Thomeer MJ, Kokkarinen J, Dalphin JC, Taylor AN. Interstitial lung diseases: an epidemiological overview. The European respiratory journal. Supplement. 2001 Sep:32():2s-16s [PubMed PMID: 11816822]
Level 2 (mid-level) evidenceTazelaar HD, Wright JL, Churg A. Desquamative interstitial pneumonia. Histopathology. 2011 Mar:58(4):509-16. doi: 10.1111/j.1365-2559.2010.03649.x. Epub 2010 Sep 21 [PubMed PMID: 20854463]
Craig PJ, Wells AU, Doffman S, Rassl D, Colby TV, Hansell DM, Du Bois RM, Nicholson AG. Desquamative interstitial pneumonia, respiratory bronchiolitis and their relationship to smoking. Histopathology. 2004 Sep:45(3):275-82 [PubMed PMID: 15330806]
Ryu JH, Myers JL, Capizzi SA, Douglas WW, Vassallo R, Decker PA. Desquamative interstitial pneumonia and respiratory bronchiolitis-associated interstitial lung disease. Chest. 2005 Jan:127(1):178-84 [PubMed PMID: 15653981]
Akira M, Yamamoto S, Hara H, Sakatani M, Ueda E. Serial computed tomographic evaluation in desquamative interstitial pneumonia. Thorax. 1997 Apr:52(4):333-7 [PubMed PMID: 9196515]
Margaritopoulos GA, Harari S, Caminati A, Antoniou KM. Smoking-related idiopathic interstitial pneumonia: A review. Respirology (Carlton, Vic.). 2016 Jan:21(1):57-64. doi: 10.1111/resp.12576. Epub 2015 Jul 2 [PubMed PMID: 26138798]
Sawata T, Bando M, Nakayama M, Mato N, Yamasawa H, Sugiyama Y. Influence of Smoking in Interstitial Pneumonia Presenting with a Non-Specific Interstitial Pneumonia Pattern. Internal medicine (Tokyo, Japan). 2016:55(20):2939-2944 [PubMed PMID: 27746429]
Johkoh T, Müller NL, Cartier Y, Kavanagh PV, Hartman TE, Akira M, Ichikado K, Ando M, Nakamura H. Idiopathic interstitial pneumonias: diagnostic accuracy of thin-section CT in 129 patients. Radiology. 1999 May:211(2):555-60 [PubMed PMID: 10228542]
Lee W, Chung WS, Hong KS, Huh J. Clinical usefulness of bronchoalveolar lavage cellular analysis and lymphocyte subsets in diffuse interstitial lung diseases. Annals of laboratory medicine. 2015 Mar:35(2):220-5. doi: 10.3343/alm.2015.35.2.220. Epub 2015 Feb 12 [PubMed PMID: 25729724]
Dias C, Mota P, Neves I, Guimarães S, Souto Moura C, Morais A. Transbronchial cryobiopsy in the diagnosis of desquamative interstitial pneumonia. Revista portuguesa de pneumologia. 2016 Sep-Oct:22(5):288-90. doi: 10.1016/j.rppnen.2016.03.006. Epub 2016 Apr 25 [PubMed PMID: 27134124]
Kligerman S, Franks TJ, Galvin JR. Clinical-Radiologic-Pathologic Correlation of Smoking-Related Diffuse Parenchymal Lung Disease. Radiologic clinics of North America. 2016 Nov:54(6):1047-1063. doi: 10.1016/j.rcl.2016.05.010. Epub 2016 Aug 22 [PubMed PMID: 27719975]
Madan R, Matalon S, Vivero M. Spectrum of Smoking-related Lung Diseases: Imaging Review and Update. Journal of thoracic imaging. 2016 Mar:31(2):78-91. doi: 10.1097/RTI.0000000000000185. Epub [PubMed PMID: 26479130]
Yousem SA, Colby TV, Gaensler EA. Respiratory bronchiolitis-associated interstitial lung disease and its relationship to desquamative interstitial pneumonia. Mayo Clinic proceedings. 1989 Nov:64(11):1373-80 [PubMed PMID: 2593722]
Hartman TE, Primack SL, Kang EY, Swensen SJ, Hansell DM, McGuinness G, Müller NL. Disease progression in usual interstitial pneumonia compared with desquamative interstitial pneumonia. Assessment with serial CT. Chest. 1996 Aug:110(2):378-82 [PubMed PMID: 8697837]
Kawabata Y, Takemura T, Hebisawa A, Sugita Y, Ogura T, Nagai S, Sakai F, Kanauchi T, Colby TV, Desquamative Interstitial Pneumonia Study Group. Desquamative interstitial pneumonia may progress to lung fibrosis as characterized radiologically. Respirology (Carlton, Vic.). 2012 Nov:17(8):1214-21. doi: 10.1111/j.1440-1843.2012.02226.x. Epub [PubMed PMID: 22805187]
Level 2 (mid-level) evidenceAubry MC, Wright JL, Myers JL. The pathology of smoking-related lung diseases. Clinics in chest medicine. 2000 Mar:21(1):11-35, vii [PubMed PMID: 10763087]
Knyazhitskiy A, Masson RG, Corkey R, Joiner J. Beneficial response to macrolide antibiotic in a patient with desquamative interstitial pneumonia refractory to corticosteroid therapy. Chest. 2008 Jul:134(1):185-7. doi: 10.1378/chest.07-2786. Epub [PubMed PMID: 18628222]
Level 3 (low-level) evidenceKatzenstein AL. Smoking-related interstitial fibrosis (SRIF), pathogenesis and treatment of usual interstitial pneumonia (UIP), and transbronchial biopsy in UIP. Modern pathology : an official journal of the United States and Canadian Academy of Pathology, Inc. 2012 Jan:25 Suppl 1():S68-78. doi: 10.1038/modpathol.2011.154. Epub [PubMed PMID: 22214972]
Rama Esendagli D, Ntiamoah P, Kupeli E, Bhardwaj A, Ghosh S, Mukhopadhyay S, Mehta AC. Recurrence of primary disease following lung transplantation. ERJ open research. 2022 Apr:8(2):. pii: 00038-2022. doi: 10.1183/23120541.00038-2022. Epub 2022 May 30 [PubMed PMID: 35651363]
Remy-Jardin M, Edme JL, Boulenguez C, Remy J, Mastora I, Sobaszek A. Longitudinal follow-up study of smoker's lung with thin-section CT in correlation with pulmonary function tests. Radiology. 2002 Jan:222(1):261-70 [PubMed PMID: 11756735]
Myers JL, Katzenstein AL. Fibroblasts in focus. American journal of respiratory and critical care medicine. 2006 Sep 15:174(6):623-4 [PubMed PMID: 16959920]
Monaghan H, Wells AU, Colby TV, du Bois RM, Hansell DM, Nicholson AG. Prognostic implications of histologic patterns in multiple surgical lung biopsies from patients with idiopathic interstitial pneumonias. Chest. 2004 Feb:125(2):522-6 [PubMed PMID: 14769733]
Level 2 (mid-level) evidenceFan LL, Dishop MK, Galambos C, Askin FB, White FV, Langston C, Liptzin DR, Kroehl ME, Deutsch GH, Young LR, Kurland G, Hagood J, Dell S, Trapnell BC, Deterding RR, Children’s Interstitial and Diffuse Lung Disease Research Network (chILDRN). Diffuse Lung Disease in Biopsied Children 2 to 18 Years of Age. Application of the chILD Classification Scheme. Annals of the American Thoracic Society. 2015 Oct:12(10):1498-505. doi: 10.1513/AnnalsATS.201501-064OC. Epub [PubMed PMID: 26291470]
Langston C, Dishop MK. Diffuse lung disease in infancy: a proposed classification applied to 259 diagnostic biopsies. Pediatric and developmental pathology : the official journal of the Society for Pediatric Pathology and the Paediatric Pathology Society. 2009 Nov-Dec:12(6):421-37. doi: 10.2350/08-11-0559.1. Epub [PubMed PMID: 19323600]
Deutsch GH, Young LR, Deterding RR, Fan LL, Dell SD, Bean JA, Brody AS, Nogee LM, Trapnell BC, Langston C, Pathology Cooperative Group, Albright EA, Askin FB, Baker P, Chou PM, Cool CM, Coventry SC, Cutz E, Davis MM, Dishop MK, Galambos C, Patterson K, Travis WD, Wert SE, White FV, ChILD Research Co-operative. Diffuse lung disease in young children: application of a novel classification scheme. American journal of respiratory and critical care medicine. 2007 Dec 1:176(11):1120-8 [PubMed PMID: 17885266]
Level 2 (mid-level) evidenceMatsuo A, Matsumoto N, Kitamura A, Tsubouchi H, Yanagi S, Nakazato M. Desquamative interstitial pneumonia complicated by diffuse alveolar haemorrhage. Respirology case reports. 2018 Feb:6(2):e00291. doi: 10.1002/rcr2.291. Epub 2017 Dec 22 [PubMed PMID: 29321935]
Level 3 (low-level) evidenceYogo Y, Oyamada Y, Ishii M, Hakuno H, Fujita A, Yamauchi T, Sawafuji M, Kobayashi K, Mukai M, Yamaguchi K. [A case of acute exacerbation of desquamative interstitial pneumonia after video-assisted thoracoscopic surgery (VATS)]. Nihon Kokyuki Gakkai zasshi = the journal of the Japanese Respiratory Society. 2003 Jun:41(6):386-91 [PubMed PMID: 12833843]
Level 3 (low-level) evidenceSánchez Varilla JM, Vázquez Martín M, García Dante H, Peñas del Castillo J, Domínguez Plata T. [Acute exacerbation of desquamative interstitial pneumonia after thoracoscopy lung biopsy]. Anales de medicina interna (Madrid, Spain : 1984). 2005 Dec:22(12):604-5 [PubMed PMID: 16523552]
Level 3 (low-level) evidenceKadoch MA, Cham MD, Beasley MB, Ward TJ, Jacobi AH, Eber CD, Padilla ML. Idiopathic interstitial pneumonias: a radiology-pathology correlation based on the revised 2013 American Thoracic Society-European Respiratory Society classification system. Current problems in diagnostic radiology. 2015 Jan-Feb:44(1):15-25. doi: 10.1067/j.cpradiol.2014.07.005. Epub 2014 Nov 11 [PubMed PMID: 25512168]
Fink L. [Interstitial lung diseases. The pattern is important]. Der Pathologe. 2014 Nov:35(6):597-605. doi: 10.1007/s00292-014-1923-1. Epub [PubMed PMID: 25319226]
Level 2 (mid-level) evidence