Introduction
Orbital hypertelorism is an abnormally increased distance between the eyes due to true lateral displacement of the orbits.[1] While hypertelorism can technically describe the increased spacing between any paired structures, it most commonly refers to orbital spacing. Hypertelorism can result from several disruptions in normal craniofacial development. Premature ossification of the lesser wings of the sphenoid bone can fix the orbits in a fetal position, preventing their normal medial migration. Additionally, failure of nasal capsule development can allow the primitive brain vesicle to expand into the space typically occupied by the capsule. Disturbances in skull base development, as occurs in craniosynostosis syndromes such as Crouzon or Apert syndromes, can also result in lateral displacement of the orbits.
In cases of orbital hypertelorism, anthropometric measurements reveal an increased inner canthal distance (ICD), outer canthal distance (OCD), and interpupillary distance (IPD), all typically exceeding the 95th percentile, or 2 standard deviations above the mean, of normative values.[2] An isolated increase in ICD without lateral orbital displacement is more accurately termed telecanthus. For this discussion, the term hypertelorism will refer to orbital hypertelorism, with telecanthus addressed in the "Differential Diagnosis" section.
A comprehensive head-to-toe dysmorphology examination is essential in all patients with hypertelorism. Additional evaluation may include genetic testing, computed tomography (CT), magnetic resonance imaging, echocardiography, renal ultrasound, and other specialty evaluations based on suspected syndromes. Treatment is typically surgical and pursued for cosmetic or functional reasons, especially in severe cases. Procedures such as box osteotomy, facial bipartition, or monobloc advancement are chosen based on orbital anatomy, maxillary arch configuration, and associated craniofacial anomalies.[3] Optimal care requires a multidisciplinary team, including craniofacial surgeons, neurosurgeons, ophthalmologists, geneticists, psychologists, and allied health professionals. Early identification, coordinated care, and personalized surgical planning are essential for improving aesthetic, functional, and psychosocial outcomes in patients with hypertelorism.
Etiology
Register For Free And Read The Full Article
 Search engine and full access to all medical articles
Search engine and full access to all medical articles- 10 free questions in your specialty
- Free CME/CE Activities
- Free daily question in your email
- Save favorite articles to your dashboard
- Emails offering discounts
Learn more about a Subscription to StatPearls Point-of-Care
Etiology
Experts believe hypertelorism is a result of an alteration in embryological facial development between the fourth and eighth week of fetal development. The frontonasal prominence is the embryological precursor of the forehead and nose. During normal development of the frontonasal prominence, lateral movement of the orbits followed by medial migration occurs. If an arrest occurs during the normal development of the frontonasal prominence, the primitive brain fills the space, preventing the normal medial migration of the orbits, causing an arrest in a lateral position.[4]
A frank encephalocele and other masses in the frontonasal prominence can produce an obvious and extreme form of hypertelorism. Additional proposed mechanisms of hypertelorism include early ossification of the lesser wings of the sphenoid bone, preventing medial movement of the orbits, and causing fixation in the fetal position. In addition, craniosynostosis syndromes that result in premature closure of cranial sutures prevent normal orbital migration and development.[1] Children with Apert syndrome, one of the most common craniosynostosis syndromes, present with hypertelorism due to the prolapse of the cribriform plate of the ethmoid bone, causing a disturbance of the cranial base formation (see Image. Craniofacial and Oral Findings in Apert Syndrome).[4] Common genes involved in the pathogenesis of craniosynostosis include fibroblast growth factor receptor 1 (FGFR1), FGFR2, FGFR3, muscle segment homeobox (MSX2), ephrin-B1 (EFNB1), and transcription factor twist homologue 1 (TWIST1).[5]
Hypertelorism is not a syndrome in and of itself but rather a physical finding in many craniofacial syndromes. When evaluating a patient with hypertelorism, clinicians must consider the proposed embryological mechanisms. When the embryonic frontonasal prominence fails to develop normally, the patient will present with median facial clefting (also often referred to as frontonasal dysplasia). Alternatively, when dysplasia of bone development is the underlying contributive factor to hypertelorism, patients will present with craniosynostosis or cranial bone abnormalities.
Syndromes associated with hypertelorism include the following:
- Craniosynostosis syndromes like Pfeiffer syndrome, Apert syndrome, Crouzon syndrome, Muenke syndrome, Jackson-Weiss syndrome, and Boston-type MSX2-related syndrome
- Bohring-Optiz syndrome
- Greig Cephalopolysyndactyly
- Noonan syndrome
- Joubert syndrome
- 4p deletion
- 9p duplication
- Tetrasomy 12p
- Trisomy 18
- 22q11 syndrome
- Multiple pterygium syndrome
- Neurofibromatosis type 1
- Aarskog syndrome
- Cat-eye syndrome
- CHARGE association
- Larsen syndrome
- Loeys-Dietz syndrome
- Wolf-Hirshorn syndrome
- Morquio syndrome
- Opitz BBB syndrome
- Pena Shokeir syndrome
- Roberts syndrome
- Skeletal dysplasias
- Hurler Syndrome [6][7]
Epidemiology
Frontonasal dysplasia or median facial clefting is rare, presenting in approximately 0.17% of all patients with clefting.[8] The incidence of clefting is approximately 1 per 700 births.[9] Craniosynostosis occurs in approximately 1 per 2000 live births, though not all patients with craniosynostosis syndromes have hypertelorism.[10] The overall estimated incidence of hypertelorism is rare, with some sources estimating the occurrence to be approximately 1 in 20,000 births.[11]
Pathophysiology
Hypertelorism occurs due to increased distance between the orbits, often resulting from developmental anomalies affecting craniofacial structures. The pathogenesis primarily involves aberrant neural crest cell migration and disrupted embryologic development of the cranial base.[12] The condition is frequently associated with midline defects, such as frontonasal dysplasia, craniosynostosis syndromes, and chromosomal abnormalities such as trisomy 18.
During normal development, the orbits form from neural crest-derived mesenchyme, contributing to facial morphogenesis. The failure of proper fusion of the frontonasal and maxillary processes or premature synostosis of cranial sutures leads to outward displacement of the orbits. Additionally, anomalies in the sphenoid and ethmoid bones contribute to the widened interorbital distance. Disruptions in signaling pathways, such as FGFR2 mutations in syndromic craniosynostosis, further support the genetic basis of hypertelorism.[6][13]
History and Physical
Isolated hypertelorism is rare. Thus, clinicians must evaluate a patient with hypertelorism for additional physical findings. Clinicians typically observe hypertelorism at birth and, in some cases, even detect it prenatally as fetal ultrasound technology becomes more advanced.[7] Hypertelorism associated with medial clefting of the face or dysplasia of nasal capsule development is more likely to be associated with other congenital anomalies. Results from a study evaluating prenatally diagnosed hypertelorism revealed karyotypic aneuploidy as the most common finding occurring in 82% of fetuses. Study results reveal that all 11 fetuses have additional anomalies of the nervous system (n = 10), skeleton (n = 6), heart (n = 5), kidneys (n = 5), or abdomen (n = 3).[14]
Hypertelorism associated with craniofacial synostosis may be syndromic or nonsyndromic. Of the craniosynostosis syndromes, patients with Apert syndrome have a sunken appearance of the midface, beaked nose, crowding of the teeth, an underdeveloped maxilla, visual difficulties secondary to shallow eye sockets, mild to moderate intellectual disability, syndactyly or polydactyly, hyperhidrosis, and cleft palate.[15] Carpenter syndrome can present with a cloverleaf skull shape, low set ears, abnormal dentition, syndactyly or polydactyly, intellectual disability, umbilical hernia, obesity, congenital heart disease, hip and spine deformities, and genital abnormalities.[16] Patients with Crouzon syndrome can present with midface hypoplasia, a beaked nose, dental anomalies, hearing loss, and normal intelligence (see Image. Crouzon Syndrome in a Patient Exhibiting Craniofacial Deformities).[17]
Ultimately, the physical examination is variable, depending on the underlying etiology of hypertelorism. Thus, when clinicians discover hypertelorism, they must conduct a comprehensive physical examination to identify additional dysmorphic features or congenital abnormalities. Evaluation of a patient with hypertelorism, or pseudohypertelorism, is not complete without a comprehensive, head-to-toe physical examination for dysmorphology. Exam findings may offer clues to an underlying syndromic diagnosis.
Physical examination may reveal the following findings:
Craniofacial Examination
- Flat or prominent nasal bridge
- Small mandible
- Flat or prominent occiput
- Metopic ridge
- Low-set ears
- Large posterior fontanelle
- Malar hypoplasia
- Anteverted nose [18]
Ear Examination
- Preauricular tags or sinus
- Large or small ears
- Asymmetric size
- Posterior rotation
- Lack of usual fold of helix [18]
Oral Examination
- Bifid uvula
- High-arched palate
- Wide alveolar ridges
- Large tongue
- Thin upper lip
- Flat philtrum [18]
Ocular Examination
- Synophrys
- Epicanthal folds
- Hypo- and hypertelorism
- Ptosis
- Short palpebral fissures
- Upward slant to palpebral fissures
- Downward slant to palpebral fissures [18]
Integumentary Examination
- Low hairline
- Frontal upsweep/aberrant hair whorl
- Alopecia of the scalp
- Extra posterior cervical skin
- A large capillary hemangioma, other than on the posterior neck
- Café au lait spots
- Hypopigmented macules [18]
Chest Examination
- Short sternum
- Depressed sternum
- Wide-set or high-located nipples
- Shield chest [18]
Abdominal and Perineal Examination
- Deep sacral dimple
- Diastasis recti (>3 cm)
- Aplasia cutis congenital
- Umbilical hernia
- Inguinal hernia
- Small testes
- Hypospadias
- Small or hypoplastic genitals [18]
Foot Examination
- Syndactyly of toes
- Overlapping toes
- Wide gap ("sandal-gap") between toes
- Prominent heel
- Broad hallux
- Hallux valgus
- Hypoplastic nails [18]
Hand Examination
- Single palmar crease
- Other unusual crease patterns
- Clinodactyly
- Duplication of the nail
- Camptodactyly
- Partial cutaneous syndactyly
- Proximally placed thumb
- Broad thumb
- Duplication of thumbnail
- Small or dysplastic nails
- Overlapping fingers
- Long fingers
- Small or large hands
- Short metacarpals [18]
Evaluation
Evaluating a patient with hypertelorism involves taking specific facial measurements. While bony interorbital distances obtained through radiographs or computed tomography (CT) scans provide the most accurate data—especially for surgical planning—these methods are not practical for the initial clinical assessment.[19] Instead, clinicians obtain precise measurements using proper techniques and anatomical landmarks (see Image. Landmarks for Measuring Intercanthal Distance, Interpupillary Distance, and Outer Canthal Distance). To perform these measurements, the patient should sit comfortably facing the examiner, with both heads level and the patient looking straight ahead. (See Table. Mean Inner Canthal Distance by Age and Table. Outer Canthal Distance by Age.) A small transparent plastic ruler is placed across the nasal bridge to measure the ICD, OCD, and IPD. These values are then compared to normative data, with hypertelorism typically defined by measurements exceeding the 95th percentile. Standardized normative value tables, including abbreviated versions, are readily available online. The following tables include an abridged version adapted from the standard normative values.[20][21]
Table. Mean Inner Canthal Distance by Age
| Age | Mean inner canthal distance by age (2 standard deviations) |
| Premature newborn | 1.6 cm (0.4) |
| Full-term newborn | 2.0 cm (0.4) |
| 1 to 6 months | 2.2 cm (0.5) |
| 7 to 12 months | 2.5 cm (0.5) |
| 13 to 18 months | 2.5 cm (0.6) |
| 19 to 24 months | 2.5 cm (0.4) |
| 25 to 30 months | 2.6 cm (0.6) |
Table. Outer Canthal Distance by Age
| Age | Outer canthal distance by age (2 standard deviations) |
| Premature newborn | 5.8 cm ( 0.7) |
| Full-term newborn | 7.0 cm ( 0.8) |
| 1 to 6 months | 7.5 cm ( 1.0) |
| 7 to 12 months | 7.8 cm ( 1.4) |
| 13 to 18 months | 8.5 cm ( 1.0) |
| 19 to 24 months | 8.2 cm ( 1.0) |
| 25 to 30 months | 8.6 cm ( 1.1) |
In adults, clinicians use the Tessier classification to differentiate hypertelorism into 3 degrees based on the ICD. The Tessler classification system is as follows:
- First degree: The ICD is 30 to 34 mm
- Second degree: The ICD is 35 to 40 mm
- Third degree: The ICD is greater than 40 mm [22]
CT scan classification or Munro and Das classification classifies hypertelorism based on the orientation of the medial orbital walls on axial scan (see Image. Munro and Das Classification Based on Computed Tomography Appearance of Orbital Medial Walls).
- Type I: Parallel medial orbital walls
- Type II: Wedge-shaped walls posteriorly
- Type III: Oval widest dimension
- Type IV: Wedge-shaped anteriorly positioned ethmoid bone, posterior to the globe [23][24]
Clinicians determine additional necessary testing based on the associated examination findings. Depending on the underlying etiology or genetic syndrome contributing to the finding of hypertelorism, additional tests may include genetic testing, an echocardiogram to evaluate for congenital heart disease, a renal ultrasound to assess for structural congenital malformations, a skeletal survey, brain magnetic resonance imaging to evaluate underlying neuronal changes, or a mass, retinal examination, audiology testing, or sleep study.
Treatment / Management
Surgical correction of hypertelorism is primarily pursued for cosmetic reasons, though it can also address functional concerns in severe cases. The procedure is complex and typically requires both intracranial and extracranial approaches. Dr Paul Tessier pioneered this surgery, showing that surgeons could reposition the orbits without compromising vision. The primary goals are to medially reposition the orbits, correct any orbital dystopia, reconstruct the nasal bridge, and remove excess soft tissue to restore facial symmetry.
Surgeons usually operate when children are between 5 and 7 years of age. Operating before age 5 can interfere with the development of the maxillary arch and dental structures and present challenges due to underdeveloped bone strength. While surgical outcomes are more predictable in adults, early intervention is often preferred to address the psychosocial impacts of visible facial differences. Coexisting craniosynostosis should undergo correction within the first year of life. Orbital osteotomies are typically paired with soft tissue adjustments to create a more natural facial appearance.
The correction of hypertelorism requires a highly individualized, multidisciplinary approach. A collaborative team—often including craniofacial surgeons, neurosurgeons, and ophthalmologists—works together to plan and execute the procedure. Advances in imaging and 3- dimensional surgical planning have significantly improved precision and outcomes. Preoperative assessments guide the surgical plan, considering factors such as the degree of orbital displacement, associated craniofacial anomalies, orbital axis, and maxillary morphology.
Surgical Approaches
Box osteotomy
A box osteotomy involves a combined medial and lateral orbital osteotomy, followed by repositioning the orbits closer to the midline. This procedure lets the surgeon medially reposition the entire bony orbit by making precise bone cuts around the orbital rim, effectively narrowing the interorbital distance. Clinicians utilize this technique in patients with a normal maxillary arch and dental occlusion, normal orbital axis, and mild to moderate hypertelorism. The size and shape of the maxillary arch are crucial; if these are normal, clinicians prefer a box osteotomy as the focus is solely on repositioning the orbits.[25]
Facial bipartition (Van der Meulen Technique)
Clinicians use the facial bipartition or the Van der Meulen technique when hypertelorism is present along with narrowing of the maxillary arch as in Apert syndrome.[26][27] The procedure medializes and rotates the orbits, and lateralizes and expands the maxillary arch. The surgical approach involves making vertical osteotomies or bone cuts on either side of the facial midline, separating the midface into 2 distinct segments. Once separated, the facial halves are rotated inwards towards the midline, allowing for the narrowing of the orbital distance and correction of the width of the maxillary arch.[28]
Monobloc advancement
Monobloc advancement involves repositioning and forward advancement of the entire front of the skull. This technique is useful for patients with severe craniofacial deformities. Osteotomies are made along the facial skeleton, detaching the midface from the skull. Then, clinicians move the entire facial structure to narrow the space between the eyes and correct other facial features.
Additional procedures
Patients may require multiple other procedures and staged surgeries. Additional procedures include segmental orbital osteotomy, U-shaped osteotomy, rhinoplasty, canthopexy, dorsal nasal augmentation, and fronto-galeal flap.[1][29]
Differential Diagnosis
Pseudohypertelorism and telecanthus are important considerations in the differential diagnosis of hypertelorism, as both can create the appearance of widely spaced eyes without actual lateral displacement of the orbits. Pseudohypertelorism is a false impression of orbital widening, often due to facial features such as a flat nasal bridge, epicanthic folds, exotropia, narrow palpebral fissures, or widely spaced eyebrows. Telecanthus, on the other hand, is defined by an increased inner canthal distance with a normal outer canthal distance and no actual change in orbital positioning. Telecanthus is frequently associated with prominent epicanthic folds and is present in various genetic syndromes, including Down syndrome, Ehlers-Danlos syndrome, Klinefelter syndrome, Turner syndrome, fetal alcohol syndrome, Cri du Chat syndrome, and Waardenburg syndrome.[30]
Differentiating true hypertelorism from these mimicking conditions is essential for accurate diagnosis and appropriate management. Unlike true hypertelorism—which involves actual lateral displacement of the bony orbits—pseudohypertelorism and telecanthus do not require surgical correction of orbital positioning. Accurate clinical evaluation helps distinguish between these entities and guides further genetic, surgical, or supportive interventions.
Prognosis
Long-term outcomes depend on the underlying cause and the presence of other abnormalities. Patients with mild, isolated hypertelorism usually have a favorable prognosis, while those with syndromic hypertelorism may experience a range of developmental challenges and health complications related to associated congenital anomalies.
Complications
Because hypertelorism is associated with genetic syndromes, affected patients may experience developmental delays, intellectual disabilities, midline clefts, encephaloceles, and agenesis of the corpus callosum, among other anomalies. Their physical appearance may cause a negative self-image and difficulty integrating with peers. Patients who undergo surgical correction are at risk for bleeding, cerebrospinal fluid leak, infection, injury to the brain, blood vessels, and cranial nerves, ophthalmic injuries, dural tears, vision loss, blindness, and ptosis. In addition, affected patients can have a recurrence of hypertelorism.[29]
Consultations
True orbital hypertelorism on an infant’s physical exam should prompt further evaluation and referral to a multidisciplinary team, including geneticists, craniofacial surgeons, and ophthalmologists. Additional specialist consultations may also be necessary for comprehensive care if a specific genetic syndrome is suspected or confirmed.
Deterrence and Patient Education
Orbital hypertelorism refers specifically to an abnormally increased distance between the orbits caused by true lateral displacement, with ICD, OCD, and IPD typically exceeding the 95th percentile of normative values. Although hypertelorism can describe the increased spacing between any paired structures, it most commonly applies to the orbits. The condition likely results from a disruption in embryologic facial development between the fourth and eighth week of gestation, particularly involving the frontonasal prominence. Arrested medial migration of the orbits during this critical period may lead to persistent lateral positioning due to the primitive brain occupying the midline space.
Healthcare professionals should educate families that hypertelorism can occur in isolation or as part of a broader genetic syndrome. A thorough examination is essential to identify any associated dysmorphic features that may suggest a syndromic diagnosis. Early referral to a multidisciplinary team—including geneticists, craniofacial surgeons, and ophthalmologists—is crucial for further evaluation and care planning if hypertelorism is suspected. Surgical correction is typically pursued for cosmetic reasons but may also address functional issues in more severe cases. Families should understand the importance of early assessment, the role of imaging and genetic testing, and the potential timing and goals of surgery. Providing supportive education and access to genetic counseling helps families make informed decisions and promotes shared, patient-centered care throughout the diagnostic and treatment process.
Enhancing Healthcare Team Outcomes
Orbital hypertelorism occurs when the orbits are abnormally far apart due to actual lateral displacement. Clinicians diagnose this condition based on anthropometric measurements—ICD, OCD, and IPD—that exceed the 95th percentile of normative values. Hypertelorism is not a diagnosis but a physical finding often seen in various craniofacial syndromes. Experts believe it results from disruptions in embryologic development, particularly involving the frontonasal prominence between the fourth and eighth weeks of gestation. Arrest in the medial migration of the orbits, often due to the expansion of the developing brain or premature ossification of cranial structures, contributes to this lateral orbital positioning.
Evaluation and management of hypertelorism requires a collaborative, multidisciplinary approach that draws on the specialized skills of clinicians, nurses, pharmacists, psychologists, occupational and physical therapists, and social workers. A thorough physical examination, advanced imaging such as CT, and additional studies—like echocardiography, renal ultrasound, and audiology testing—may be indicated depending on the suspected underlying condition. Surgical correction, pursued primarily for cosmetic reasons but occasionally for functional indications, is complex and often delayed until ages 5 to 7 to avoid interfering with craniofacial growth. Techniques include box osteotomy, facial bipartition, and monobloc advancement, each tailored to the individual’s anatomy and associated anomalies. Advances in 3 dimensional imaging and preoperative planning have significantly improved surgical precision and outcomes. An interprofessional team must ensure comprehensive assessment, coordinated care, and optimal functional and aesthetic results.
Advanced clinicians must possess strong diagnostic skills to recognize hypertelorism as a clinical finding, understand its embryologic basis, and evaluate for potential syndromic associations. They aid in initiating appropriate referrals for genetic evaluation, imaging, and surgical consultation while guiding families through the diagnostic and treatment process with empathy and clarity. Nurses are vital in providing patient education, supporting families through the often complex care process, and helping coordinate follow-up appointments and preoperative or postoperative care. Their involvement ensures continuity and reinforces the care plan.
Pharmacists contribute by reviewing perioperative medications, managing anesthesia-related risks, and ensuring safe medication use during recovery, particularly in syndromic patients with comorbid conditions.Interprofessional communication is essential in coordinating evaluations, diagnostic testing, and treatment planning. Ensuring all aspects of care are appropriately addressed requires regular case discussions among healthcare team members. Using shared electronic health records, team huddles, and care conferences helps align goals and prevent gaps in care.
A well-defined strategy includes early diagnosis, comprehensive dysmorphology assessment, and individualized surgical planning when indicated. Coordinated care ensures timely intervention, improves patient safety, and promotes optimal developmental and psychosocial outcomes. Through teamwork and shared expertise, healthcare professionals can deliver patient-centered, high-quality care tailored to the unique needs of individuals with hypertelorism.
Media
(Click Image to Enlarge)
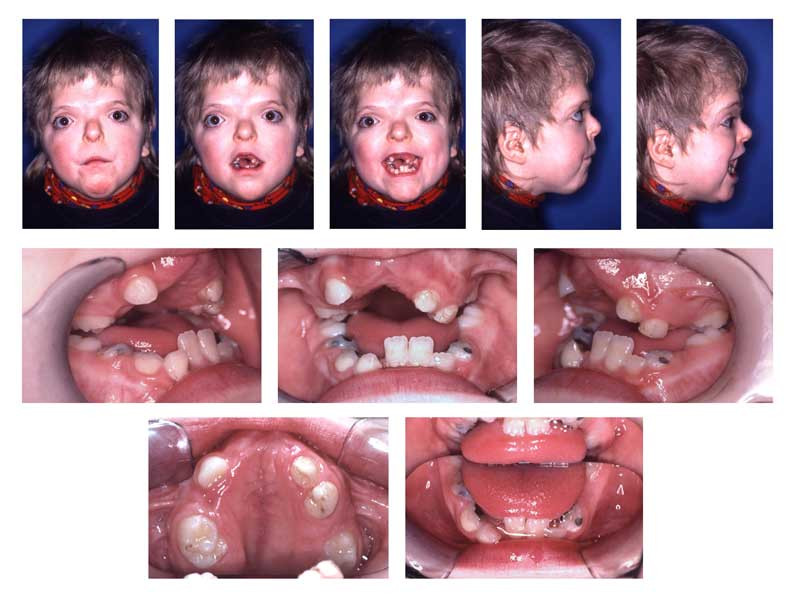
Craniofacial and Oral Findings in Apert Syndrome. The image depicts hypertelorism, vertical excess of the lower one-third of the face, a trapezoidal upper lip, and difficulty achieving forced lip closure. Intraoral changes are delayed dentition, crowding, Angle Class III malocclusion with dental compensation through lower incisor retrusion, and a circular open bite with a unilateral crossbite.
Filip, Public Domain, via Wikimedia Commons
{kind=link}
(Click Image to Enlarge)
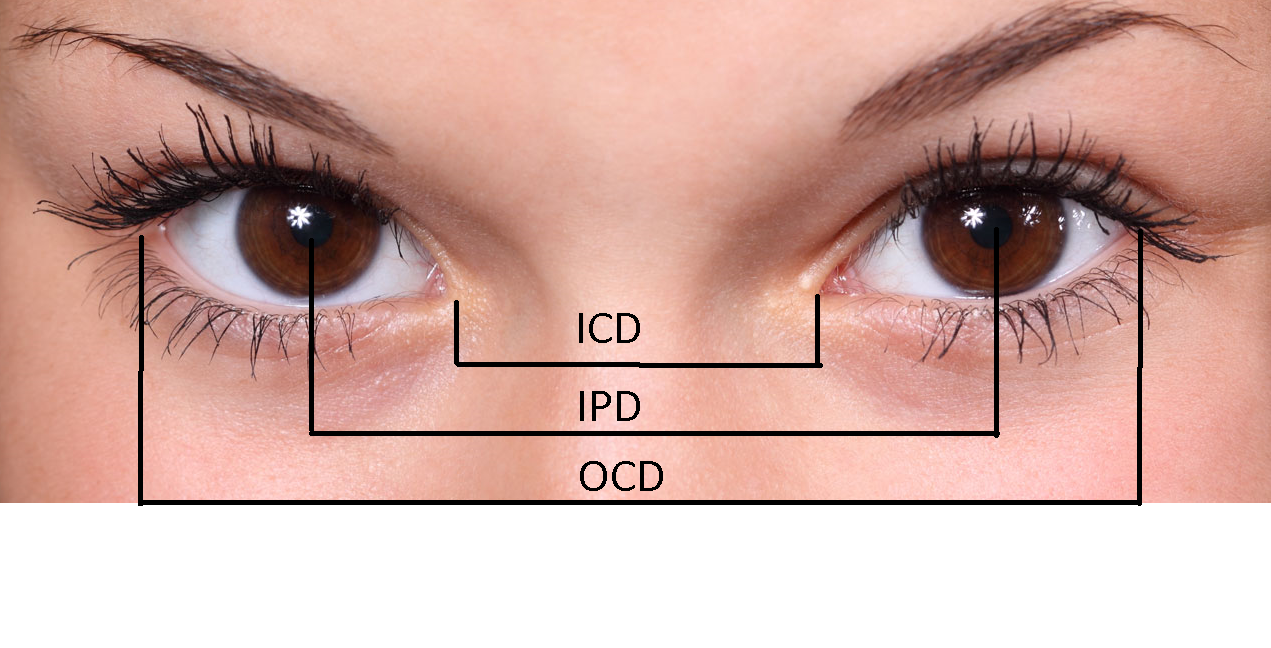
Landmarks for Measuring Intercanthal Distance, Interpupillary Distance, and Outer Canthal Distance. Evaluating a patient with hypertelorism involves taking specific facial measurements; clinicians should obtain precise measurements using proper techniques and anatomical landmarks.
Contributed by A Shillington, DO
(Click Image to Enlarge)

Crouzon Syndrome in a Patient Exhibiting Craniofacial Deformities. Patients with Crouzon syndrome can present with midface hypoplasia, a beaked nose, dental anomalies, hearing loss, and normal intelligence.
Octave Crouzon, Public Domain, via Wikimedia Commons
(Click Image to Enlarge)

Munro and Das Classification Based on Computed Tomography Appearance of Orbital Medial Walls. Computed tomography scan classification, or Munro and Das classification, classifies hypertelorism based on the orientation of the medial orbital walls on axial scan.
Contributed by K Tripathy, MD
References
Sharma RK. Hypertelorism. Indian journal of plastic surgery : official publication of the Association of Plastic Surgeons of India. 2014 Sep-Dec:47(3):284-92. doi: 10.4103/0970-0358.146572. Epub [PubMed PMID: 25593412]
Burns NS, Iyer RS, Robinson AJ, Chapman T. Diagnostic imaging of fetal and pediatric orbital abnormalities. AJR. American journal of roentgenology. 2013 Dec:201(6):W797-808. doi: 10.2214/AJR.13.10949. Epub [PubMed PMID: 24261386]
Tashima A, Brady C. Orbital Hypertelorism. Clinics in plastic surgery. 2025 Apr:52(2):209-217. doi: 10.1016/j.cps.2024.10.004. Epub 2024 Nov 7 [PubMed PMID: 39986883]
Cohen MM Jr, Richieri-Costa A, Guion-Almeida ML, Saavedra D. Hypertelorism: interorbital growth, measurements, and pathogenetic considerations. International journal of oral and maxillofacial surgery. 1995 Dec:24(6):387-95 [PubMed PMID: 8636632]
Level 3 (low-level) evidenceJohnson D, Wilkie AO. Craniosynostosis. European journal of human genetics : EJHG. 2011 Apr:19(4):369-76. doi: 10.1038/ejhg.2010.235. Epub 2011 Jan 19 [PubMed PMID: 21248745]
Adam MP, Feldman J, Mirzaa GM, Pagon RA, Wallace SE, Amemiya A, Wenger T, Miller D, Evans K. FGFR Craniosynostosis Syndromes Overview. GeneReviews(®). 1993:(): [PubMed PMID: 20301628]
Level 3 (low-level) evidenceSociety for Maternal-Fetal Medicine, Benacerraf BR, Bromley BS, Jelin AC. Hypertelorism. American journal of obstetrics and gynecology. 2019 Nov:221(5):B18-B19. doi: 10.1016/j.ajog.2019.08.053. Epub [PubMed PMID: 31679589]
Dube G, Jain S. Prevalence of True Median Cleft of Upper Lip as Reflected from a Small Central Indian Population: Attempt to Report and Review the True Median Cleft of Upper Lip. Journal of maxillofacial and oral surgery. 2018 Dec:17(4):508-513. doi: 10.1007/s12663-017-1072-1. Epub 2017 Dec 16 [PubMed PMID: 30344394]
Bernheim N, Georges M, Malevez C, De Mey A, Mansbach A. Embryology and epidemiology of cleft lip and palate. B-ENT. 2006:2 Suppl 4():11-9 [PubMed PMID: 17366840]
Betances EM,Mendez MD,M Das J, Craniosynostosis 2020 Jan; [PubMed PMID: 31335086]
Kimonis V, Gold JA, Hoffman TL, Panchal J, Boyadjiev SA. Genetics of craniosynostosis. Seminars in pediatric neurology. 2007 Sep:14(3):150-61 [PubMed PMID: 17980312]
Siismets EM, Hatch NE. Cranial Neural Crest Cells and Their Role in the Pathogenesis of Craniofacial Anomalies and Coronal Craniosynostosis. Journal of developmental biology. 2020 Sep 9:8(3):. doi: 10.3390/jdb8030018. Epub 2020 Sep 9 [PubMed PMID: 32916911]
Yapijakis C, Pachis N, Sotiriadou T, Vaila C, Michopoulou V, Vassiliou S. Molecular Mechanisms Involved in Craniosynostosis. In vivo (Athens, Greece). 2023 Jan-Feb:37(1):36-46. doi: 10.21873/invivo.13052. Epub [PubMed PMID: 36593018]
Bergé SJ, Plath H, Van de Vondel PT, Appel T, Niederhagen B, Von Lindern JJ, Reich RH, Hansmann M. Fetal cleft lip and palate: sonographic diagnosis, chromosomal abnormalities, associated anomalies and postnatal outcome in 70 fetuses. Ultrasound in obstetrics & gynecology : the official journal of the International Society of Ultrasound in Obstetrics and Gynecology. 2001 Nov:18(5):422-31 [PubMed PMID: 11844159]
Level 2 (mid-level) evidenceCha BK, Choi DS, Jang IS, Yook HT, Lee SY, Lee SS, Lee SK. Aberrant growth of the anterior cranial base relevant to severe midface hypoplasia of Apert syndrome. Maxillofacial plastic and reconstructive surgery. 2018 Dec:40(1):40. doi: 10.1186/s40902-018-0179-8. Epub 2018 Dec 12 [PubMed PMID: 30591916]
Begum S, Khatun N, Rayhan SM, Rahman SA. Carpenter syndrome: a case report. Mymensingh medical journal : MMJ. 2012 Jul:21(3):547-9 [PubMed PMID: 22828559]
Level 3 (low-level) evidenceBowling EL, Burstein FD. Crouzon syndrome. Optometry (St. Louis, Mo.). 2006 May:77(5):217-22 [PubMed PMID: 16651211]
Level 3 (low-level) evidenceMARDEN PM, SMITH DW, MCDONALD MJ. CONGENITAL ANOMALIES IN THE NEWBORN INFANT, INCLUDING MINOR VARIATIONS. A STUDY OF 4,412 BABIES BY SURFACE EXAMINATION FOR ANOMALIES AND BUCCAL SMEAR FOR SEX CHROMATIN. The Journal of pediatrics. 1964 Mar:64():357-71 [PubMed PMID: 14130709]
Mafee MF, Pruzansky S, Corrales MM, Phatak MG, Valvassori GE, Dobben GD, Capek V. CT in the evaluation of the orbit and the bony interorbital distance. AJNR. American journal of neuroradiology. 1986 Mar-Apr:7(2):265-9 [PubMed PMID: 3082161]
Laestadius ND, Aase JM, Smith DW. Normal inner canthal and outer orbital dimensions. The Journal of pediatrics. 1969 Mar:74(3):465-8 [PubMed PMID: 5764779]
Feingold M, Bossert WH. Normal values for selected physical parameters: an aid to syndrome delineation. Birth defects original article series. 1974:10(13):1-16 [PubMed PMID: 4470702]
Winters R. Tessier Clefts and Hypertelorism. Facial plastic surgery clinics of North America. 2016 Nov:24(4):545-558. doi: 10.1016/j.fsc.2016.06.013. Epub [PubMed PMID: 27712820]
Fearon JA, Bartlett SP, Whitaker LA. The skeletal treatment of orbital hypertelorism. Neurosurgery clinics of North America. 1991 Jul:2(3):673-81 [PubMed PMID: 1821313]
Munro IR, Das SK. Improving results in orbital hypertelorism correction. Annals of plastic surgery. 1979 Jun:2(6):499-507 [PubMed PMID: 543618]
de Sousa BC, Ferreira-Pinto PHC, Ferreira DBCO, Bastos EP, Junior MLLA, Dias BSB, Schneider T, Claro V, Cintra HPL, Parise M, Correa EM, Cruz TZ, da Silva WN, Nigri F. Isolated hypertelorism: Late surgical correction using the box osteotomy technique. Surgical neurology international. 2024:15():145. doi: 10.25259/SNI_1029_2023. Epub 2024 Apr 26 [PubMed PMID: 38741988]
Batut C, Joly A, Travers N, Guichard B, Paré A, Laure B. Surgical treatment of orbital hypertelorism: Historical evolution and development prospects. Journal of cranio-maxillo-facial surgery : official publication of the European Association for Cranio-Maxillo-Facial Surgery. 2019 Nov:47(11):1712-1719. doi: 10.1016/j.jcms.2019.07.002. Epub 2019 Jul 13 [PubMed PMID: 31519384]
van der Meulen JC. Medial faciotomy. British journal of plastic surgery. 1979 Oct:32(4):339-42 [PubMed PMID: 534804]
Balaji SM. Modified facial bipartition. Annals of maxillofacial surgery. 2012 Jul:2(2):170-3. doi: 10.4103/2231-0746.101348. Epub [PubMed PMID: 23483335]
Shakir S, Hoppe IC, Taylor JA. State-of-the-Art Hypertelorism Management. Clinics in plastic surgery. 2019 Apr:46(2):185-195. doi: 10.1016/j.cps.2018.11.004. Epub 2019 Jan 9 [PubMed PMID: 30851750]
Dollfus H, Verloes A. Dysmorphology and the orbital region: a practical clinical approach. Survey of ophthalmology. 2004 Nov-Dec:49(6):547-61 [PubMed PMID: 15530943]
Level 3 (low-level) evidence