Introduction
Lennox-Gastaut syndrome (LGS) is a lifelong, severe developmental epileptic encephalopathy characterized by treatment-resistant epilepsy, intellectual disability, and multiple seizure types—most notably tonic, atypical absence, and atonic seizures.[1][2][3][4] A hallmark electroencephalogram (EEG) feature includes interictal slow spike-wave patterns, often accompanied by generalized paroxysmal fast activity during sleep. LGS can arise from a variety of underlying causes, such as genetic mutations, cortical malformations, brain tumors, neurocutaneous disorders such as tuberous sclerosis complex, hypoxic-ischemic injury, meningitis, or head trauma. However, in approximately 40% of cases, no identifiable cause is found.[2]
LGS is diagnosed by the presence of tonic seizures, along with at least one additional type of seizure, with onset before the age of 18. The diagnosis also requires the characteristic EEG findings, coupled with a long-term history of treatment-resistant epilepsy and severe intellectual disability. Sleep EEG is particularly useful in identifying key patterns of seizure activity. Magnetic resonance imaging (MRI) of the brain and genetic testing are crucial for determining the underlying cause. Seizure management is often challenging in patients with LGS and typically involves a combination of antiseizure medications. Despite treatment, most patients continue to experience seizures and require ongoing multidisciplinary care.[5][6]
Etiology
Register For Free And Read The Full Article
 Search engine and full access to all medical articles
Search engine and full access to all medical articles- 10 free questions in your specialty
- Free CME/CE Activities
- Free daily question in your email
- Save favorite articles to your dashboard
- Emails offering discounts
Learn more about a Subscription to StatPearls Point-of-Care
Etiology
LGS can result from various causes. Secondary LGS accounts for approximately 60% of cases and is associated with identifiable underlying brain pathology, usually from diffuse cerebral injury.[7] Common causes include tuberous sclerosis, central nervous system infections (such as encephalitis or meningitis), frontal lobe injuries, birth trauma, metabolic disorders, and developmental brain malformations. About 25% of infants who go on to develop LGS have a prior history of infantile spasms, which is often associated with a more severe disease course. These children typically face a poorer prognosis and higher mortality rates.[8][9]
Alternatively, idiopathic or cryptogenic LGS refers to cases without an identifiable underlying cause. However, recent research suggests that many of these children may have underlying genetic disorders or de novo mutations in genes such as SCN1A (sodium voltage-gated channel alpha subunit-1), GABRB3 (gamma-aminobutyric acid receptor beta-3), ALG13 (UDP-N-acetylglucosaminyltransferase subunit), and CHD2 (chromodomain helicase DNA-binding protein-2). While these discoveries are promising, the precise role of these genetic alterations in the pathogenesis of LGS remains unclear.[9][10][11][12][13]
Epidemiology
A recent study estimates the incidence of LGS to be between 14.5 and 28.0 per 100,000 individuals, with a prevalence ranging from 5.8 to 60.8 per 100,000 individuals.[6][14] Additional research suggests that LGS accounts for 1% to 2% of all epilepsy cases, 2% to 5% of childhood epilepsies, and 10% of epilepsy cases in children aged 5 or younger.[6][7][15][16][17][18][19][20] LGS is slightly more common in males, with no evidence of an ethnic predisposition.[17]
Pathophysiology
The exact pathophysiology underlying LGS remains unclear. The condition is associated with abnormal brain activity and disrupted neural connectivity, often linked to structural or genetic abnormalities. Given the typical early age of onset, it is believed that the heightened susceptibility of the immature brain contributes to the development of LGS. Interestingly, although many patients have identifiable brain lesions on MRI, those without detectable lesions often exhibit similar EEG patterns. Moreover, individuals with the same underlying pathology, such as tuberous sclerosis, may develop LGS, epileptic spasms, or focal epilepsy. These findings suggest that structural abnormalities alone do not fully explain the pathogenesis of LGS.
EEG, functional MRI, and deep brain stimulation studies suggest that the thalamus and pons play a crucial role in facilitating seizure activity in LGS.[21] Stimulation of the centromedian nucleus of the thalamus has been associated with a reduction of up to 80% in generalized seizures in some patients with LGS. However, as seizures may resolve following the removal of cortical lesions in certain individuals, the thalamus is thought to act more as a synchronizer and amplifier of epileptic activity rather than its primary source of origin. Similarly, the pons is implicated in the expression of tonic seizures, but like the thalamus, it is not considered the origin of epileptic activity in LGS.
These findings support the concept of epilepsy as a disorder of cerebral networks, involving dynamic interactions between cortical and subcortical structures. The primary cortical regions involved are the frontal and parietal lobes, which activate the cerebellum, thalamus, and pons. The primary motor and sensory cortical structures are not involved in this process. When identified, brain lesions associated with LGS interact chronically with these brain networks.[2][22] Additional theories propose that LGS may be an epileptic disorder of the reticulo-thalamo-cortical system, characterized by self-sustained epilepsy resulting from activation and epileptogenesis within the reticular activating system between the midbrain and the thalamus.[21]
History and Physical
LGS is defined by a triad of multiple seizure types, characteristic EEG findings, and intellectual disabilities.[3] Recognizing and diagnosing LGS can be challenging, as these hallmark clinical and EEG findings may be absent, particularly in the early stages of the disease. Seizures typically begin in early childhood, usually before the age of 8, with a peak onset between the ages of 3 and 5.[15] In some cases, seizures start before the age of 1 and later progress to LGS, often evolving from other epilepsy syndromes such as infantile epileptic spasms syndrome, West syndrome, or epileptic spasms. Patients with LGS experience multiple seizure types, with tonic and atypical absence seizures being particularly common.
Additional seizure types seen in LGS include atonic (drop attacks), focal impaired awareness, generalized tonic-clonic, myoclonic seizures, nonconvulsive status epilepticus, and epileptic spasms. Tonic seizures are characterized by sustained muscle contractions that involve the limbs and axial muscles, typically lasting from a few seconds to several minutes. They are a hallmark of LGS. These seizures most commonly occur during sleep but may also present during wakefulness, often resulting in sudden falls or “drop attacks.” The presence of tonic seizures is essential for establishing a diagnosis of LGS.[23]
Atypical absence seizures are the second most common subtype of seizure present in patients with LGS. Affected patients have episodes involving impaired awareness that typically begin and end gradually, making them difficult to recognize. Atonic seizures, observed in 10% to 56% of cases, cause a sudden loss of axial muscle tone, often resulting in drop attacks, which lead to recurrent falls and injuries. Controlling atonic seizures is a key priority in the treatment of LGS due to the significant risk of injury from recurrent falls.
Additionally, most patients experience nonconvulsive status epilepticus at some point after diagnosis, which further contributes to developmental and cognitive delays.[3] Symptoms may include dizziness, staring spells, apathy, stupor, and unresponsiveness. Identifying specific seizure types can be challenging due to the high frequency and variability of daily seizure episodes.[3][23] Moreover, seizure types and frequency often evolve over time. In such cases, continuous or video EEG monitoring is valuable for accurately classifying seizure activity.[24]
Children with LGS typically exhibit normal physical growth, and some may have normal neurodevelopment before the onset of seizures. However, many children may show signs of neurodevelopmental delays even before the onset of seizures. Common behavioral and developmental concerns in children with LGS include hyperactivity, aggression, autism spectrum disorder, and sleep disturbances.[25][26] Most affected children experience a developmental plateau or regression in key milestones, with decline typically worsening as they age.[3][23][27] Approximately 20% of children maintain normal memory and cognitive function, although they may experience delays in information processing.
Evaluation
The evaluation of LGS begins with a comprehensive review of the patient's pregnancy, birth, medical, cognitive, behavioral, developmental, and seizure history. Patients should undergo a comprehensive physical examination. Diagnostic laboratory tests include a complete blood count (CBC) with differential, serum electrolytes, glucose, calcium, magnesium, renal and liver function tests, urinalysis, toxicology screen, serum ammonia, lactic acid, serum amino acids, acylcarnitine profile, and urine organic acids. The diagnosis of LGS requires an interictal EEG that reveals a generalized slow spike-wave pattern, with a frequency of 2.5 Hz or less, and the highest amplitude in the frontal lobe region (see Image. Electroencephalogram in Lennox-Gastaut Syndrome).
In addition, the EEG should reveal paroxysmal fast activity, characterized by brief bursts of fast, rhythmic electrical activity, typically ranging from 10 to 30 Hz.[3][23][28] Clinicians should perform both a sleep and awake EEG, as these changes are often observed exclusively during sleep.[3][23] A video EEG may also be helpful in capturing and characterizing the different seizure types.
A brain MRI is necessary for evaluating structural brain lesions, although it is not required to establish the diagnosis of LGS. Genetic testing should include an epilepsy gene panel and a chromosomal microarray. Clinicians should consider an ECG if there are concerns about a potential cardiac cause of the child’s episodes. If neuroimaging has ruled out a space-occupying lesion, a lumbar puncture should be performed when there is suspicion of an acute infectious process.
Diagnostic Criteria for Lennox-Gastaut Syndrome
The International League Against Epilepsy (ILAE) outlines mandatory, exclusionary, and alert criteria for the diagnosis of LGS, as mentioned below.
Mandatory criteria
- Presence of tonic seizures.
- At least one additional seizure type, such as atypical absence, atonic, myoclonic, focal impaired awareness, generalized tonic-clonic, nonconvulsive status epilepticus, or epileptic spasms.
- EEG showing generalized slow spike-and-wave complexes of 2.5 Hz or less (findings from prior EEGs are acceptable).
- EEG showing generalized paroxysmal fast activity during sleep (findings from prior EEGs are acceptable).
- Symptom onset in individuals aged 18 or younger.
- Treatment-resistant epilepsy accompanied by mild-to-severe intellectual disability.[26]
Exclutionary criteria
- The diagnosis of LGS is excluded if the EEG shows persistent focal abnormalities without a generalized spike-and-wave pattern.[26]
Alert criteria
The below alert criteria should prompt clinicians to exercise caution when considering a diagnosis of LGS.
- The seizure onset age of 8 or older.
- EEG showing an exaggerated photic response at low frequencies, which may indicate late-infantile neuronal ceroid lipofuscinosis (CLN2 disease)—a rare, rapidly progressive neurodegenerative disorder caused by tripeptidyl peptidase-1 deficiency, leading to the accumulation of waste products in the brain and other organs.[26]
Treatment / Management
The management of LGS requires a comprehensive, multidisciplinary, and individualized approach throughout the patient’s life. This approach should address the patient’s medical, social, psychological, and educational needs, as well as those of their family and caregivers.[29] Medical treatment for LGS primarily focuses on seizure control and involves a combination of medical, dietary, and surgical interventions.[30][31] Effective seizure management can lead to improvements in cognition, mood, alertness, and overall quality of life.[32](B3)
Medical Management
Medications are essential in the treatment of LGS, but managing seizures remains challenging due to their treatment resistance, the variety of seizure types, and the need for combination therapy. Clinicians often prioritize controlling tonic and atonic seizures because of their association with sudden falls and injuries. However, managing one type of seizure with a specific medication can sometimes worsen another, complicating seizure management. For example, sodium channel blockers such as phenytoin and carbamazepine can exacerbate tonic, myoclonic, and drop attacks, highlighting the importance of accurately identifying seizure types for effective treatment.
Valproate, lamotrigine, rufinamide, topiramate, felbamate, benzodiazepines, and cannabidiol oral solution are all approved treatments for LGS.[33][34][35] Valproate, often used in combination with lamotrigine or clobazam, is considered a first-line therapy.[31] A tailored, seizure-specific approach is essential for effective management and minimizing complications.(A1)
- Valproate: This is effective in treating multiple seizure types and is often used as a first-line medication in the management of LGS. Although it may be used as monotherapy initially, most patients ultimately require combination therapy for optimal seizure control.[31] Due to its teratogenicity, valproate is not recommended for patients who are pregnant or of childbearing potential unless no suitable alternatives are available.
- Lamotrigine: This medication is effective for treating tonic-clonic seizures and is approved for use in children aged 2 or older. Additionally, lamotrigine may also offer benefits in improving mood, behavior, and speech in certain patients.[31][33] (A1)
- Clobazam: This drug is a long-acting benzodiazepine used as adjunctive therapy for treating seizures in patients with LGS aged 2 or older. Clobazam is especially effective in reducing atypical absence seizures.[36][37]
- Felbamate: This drug primarily treats atonic and tonic-clonic seizures. Although it was one of the first medications approved for LGS, felbamate is generally reserved for cases where other treatments have failed due to the risk of severe adverse effects.[38][39][40] (A1)
- Rufinamide: This medication is effective for treating LGS in children aged 4 or older, as well as in adults. Rufinamide is effective against atonic and tonic-clonic seizures.[41] A distinguishing feature of rufinamide is that it typically does not exacerbate other types of seizures.
- Topiramate: This drug is effective in treating tonic-clonic seizures and is also approved by the US Food and Drug Administration (FDA) as an add-on therapy for managing seizures associated with LGS in children older than 2.[31][34] (A1)
- Cannabidiol oral solution: This is the most recent FDA-approved treatment for LGS in children aged 2 or older, and it is particularly effective in reducing drop attacks, while also providing benefits for other seizure types.[42][43][44] (B3)
- Vigabatrin, zonisamide, ethosuximide, clonazepam, and levetiracetam: Although these medications have also been used, their efficacy in this syndrome remains poorly studied.[31]
- Phenytoin, carbamazepine, oxcarbazepine, gabapentin, lacosamide, and phenobarbital: These medications are not used in LGS as they can exacerbate certain seizure types associated with the condition.[45] (B2)
- Corticosteroid and intravenous immunoglobulin: These treatments may reduce seizure frequency, but their effectiveness is not supported by rigorous clinical studies.[46][47]
Clinicians should limit medication regimens to no more than 2 drugs. When adding a new antiseizure therapy to a patient already on 2 medications, one of the older medications should be gradually tapered off, as studies show no benefit from chronic drug regimens involving more than 2 medications.
Dietary Management
Seizures in LGS are often refractory to medical treatments, making dietary modifications an important next step. These modifications, studied in both children and adults, can help reduce seizure frequency and potentially decrease medication dosages. Various diets, including the ketogenic diet, modified Atkins diet, and low-glycemic-index diet, have demonstrated some effectiveness.[48][49][50] A case series found a 50% reduction in seizure activity in approximately 50% of children after 6 months on a ketogenic diet, with an additional 23% of children experiencing a 90% reduction in seizure activity.[51](B2)
Surgical Management
Surgical options may be considered for patients who do not respond to medical and dietary therapies.[52] Specifically, surgical intervention is typically explored when the first two seizure medications fail. These options may include vagus nerve stimulation (VNS) or brain surgery.[50]
Vagus nerve stimulation: VNS is typically used in combination with medical therapy and is especially effective for atonic and tonic seizures.[52] VNS can lead to an approximate 50% reduction in seizure frequency. Notably, unlike many antiseizure medications, the benefits of VNS often increase over time and may also contribute to improvements in mood and behavior.
Brain surgery: Surgical options for managing LGS include resection, disconnection procedures such as corpus callosotomy, and, in select cases, hemispherectomy. Although LGS was historically considered a generalized epilepsy syndrome, which made patients poor candidates for surgery, individuals with secondary LGS may have a localized, resectable lesion, such as a tuber, tumor, or cortical malformation, that serves as the seizure focus and could benefit from surgical removal.
Because seizures in LGS often involve both hemispheres, corpus callosotomy—a procedure that severs the connections between the 2 sides of the brain—can help limit seizure spread. This approach is particularly effective in controlling atonic, tonic, and tonic-clonic seizures.[53] Early callosotomy is especially beneficial for patients experiencing frequent drop attacks that result in serious injuries or loss of mobility. In some cases, clinicians may perform a partial or anterior corpus callosotomy to reduce risks while still achieving clinical benefit.[54]
Differential Diagnosis
As the symptoms of LGS often evolve over time, establishing a definitive diagnosis may take years of clinical follow-up.[55] The differential diagnoses for LGS include:
- Dravet syndrome
- Epilepsy with myoclonic-atonic seizures
- Atypical benign focal epilepsy of childhood
- Developmental and epileptic encephalopathy with spike-wave activation in sleep
- Pseudo-Lennox-syndrome
- Ring chromosome 20 syndrome
- Frontal lobe epilepsy
- Neuronal ceroid lipofuscinosis
- Sialidosis types I and II
- Niemann-Pick disease type C
- Gaucher disease
- Progressive myoclonic epilepsy
- Epileptic spasms
- Infantile spasms
- Infantile epileptic spasms syndrome
- West syndrome [56][57][58][59][60][61]
Prognosis
The overall prognosis for patients with LGS is poor and largely depends on the degree of seizure control. The mortality rate ranges from 3% to 7% within the first 8 to 10 years following diagnosis, primarily due to brain injuries or accidents, with an overall mortality rate of approximately 5%.[62] Despite the challenges, studies indicate that individuals with LGS can live into their 50s or beyond. Patients with a history of infantile spasms or West syndrome often experience poorer seizure control, which is associated with more severe cognitive impairment.[7][8] Additionally, sudden unexpected death in epilepsy may be more common in LGS due to the high frequency of seizures.[6][32][63]
Complications
The complications associated with LGS primarily arise from the challenges in achieving adequate seizure control. Additionally, the adverse effects of medications can contribute to the overall risk. Common complications include:
- Aspiration with pneumonia
- Status epilepticus
- Sudden unexpected death in epilepsy
- Accidents and injury-related mortality
- Severe intellectual and behavioral impairments, often necessitating placement in long-term care facilities
- Traumatic brain injury
- Fatal aplastic anemia due to felbamate
- Liver failure associated with felbamate
- Cardiac valve injury due to fenfluramine
- Pulmonary hypertension linked to fenfluramine
- Metabolic acidosis induced by topiramate
- Nephrolithiasis due to topiramate
- Hemophagocytic lymphohistiocytosis associated with lamotrigine
- Drug reaction with eosinophilia and systemic symptoms (DRESS) due to lamotrigine [59][64][65]
Deterrence and Patient Education
LGS is a lifelong, severe developmental and epileptic encephalopathy characterized by treatment-resistant epilepsy, intellectual disabilities, and multiple seizure types—most notably tonic, atypical absence, and atonic seizures. Because LGS can result from diverse underlying causes, including genetic mutations and cortical malformations, a thorough diagnostic evaluation—such as EEG, brain MRI, and genetic testing—is essential for identifying potential etiologies.
Healthcare professionals should provide early education to caregivers and families about the chronic nature of Lennox-Gastaut syndrome (LGS), its typical clinical presentation, and the importance of promptly seeking medical attention when seizure activity occurs. Clinicians should also counsel families on the long-term implications, including the high likelihood of treatment-resistant seizures and progressive cognitive decline. Notably, it is essential for patients, caregivers, and families to understand the increased risks of injuries, aspiration, drowning, and sudden unexpected death in epilepsy, and to take necessary steps to create the safest possible environment.
Early intervention, multidisciplinary care, and adherence to seizure safety practices are essential to reducing complications in patients with LGS. Patient education should focus on setting realistic expectations, encouraging routine follow-up, and promoting coordination with specialists such as neurologists, geneticists, therapists, and social workers. With appropriate support, individuals with LGS can live into adulthood, and continued caregiver involvement and education are crucial for enhancing quality of life and achieving better long-term outcomes.
Pearls and Other Issues
International Lennox-Gastaut Syndrome (LGS) Awareness Day is observed annually on November 1st.
Enhancing Healthcare Team Outcomes
LGS is a lifelong, severe developmental epileptic encephalopathy characterized by treatment-resistant epilepsy, intellectual disability, and multiple seizure types—most notably tonic, atypical absence, and atonic seizures. A hallmark EEG finding is an interictal slow spike-wave pattern, often accompanied by generalized paroxysmal fast activity during sleep. LGS can result from various etiologies, including genetic mutations, cortical malformations, tumors, hypoxic-ischemic injury, meningitis, or head trauma. However, in approximately 40% of cases, no identifiable cause is found. A diagnostic evaluation should include a brain MRI and genetic testing to help determine the underlying etiology. Management is complex and typically involves multiple antiseizure medications, as well as long-term, coordinated care from a multidisciplinary team.
Physicians and advanced practitioners use their clinical expertise to accurately diagnose LGS, determine appropriate diagnostic tests, prescribe antiseizure medications, and manage potential complications. Nurses play a vital role in educating patients and caregivers about the progression, complications, and management of LGS, ensuring early recognition of symptoms and a prompt response to adverse events. They also act as essential communicators, relaying patient and caregiver concerns to the healthcare team to help develop individualized care plans. Pharmacists are key members of the care team, particularly in managing the complex medication regimens often used in LGS. They assist with treatment planning by recommending appropriate medication combinations and dosages and ensure patient safety by monitoring for potential adverse effects and drug interactions that could impact the effectiveness of antiepileptic therapies.
Additionally, most patients and caregivers benefit from the involvement of social workers, psychologists, psychiatric specialists, and rehabilitation professionals, including physical, occupational, and speech therapists, to support functional independence and enhance quality of life. An interprofessional, collaborative approach is essential for timely diagnosis, treatment, education, and ongoing support for individuals with LGS. Coordinated efforts across disciplines improve care delivery, facilitate early intervention, and contribute to better clinical outcomes and overall patient well-being.
Media
(Click Image to Enlarge)
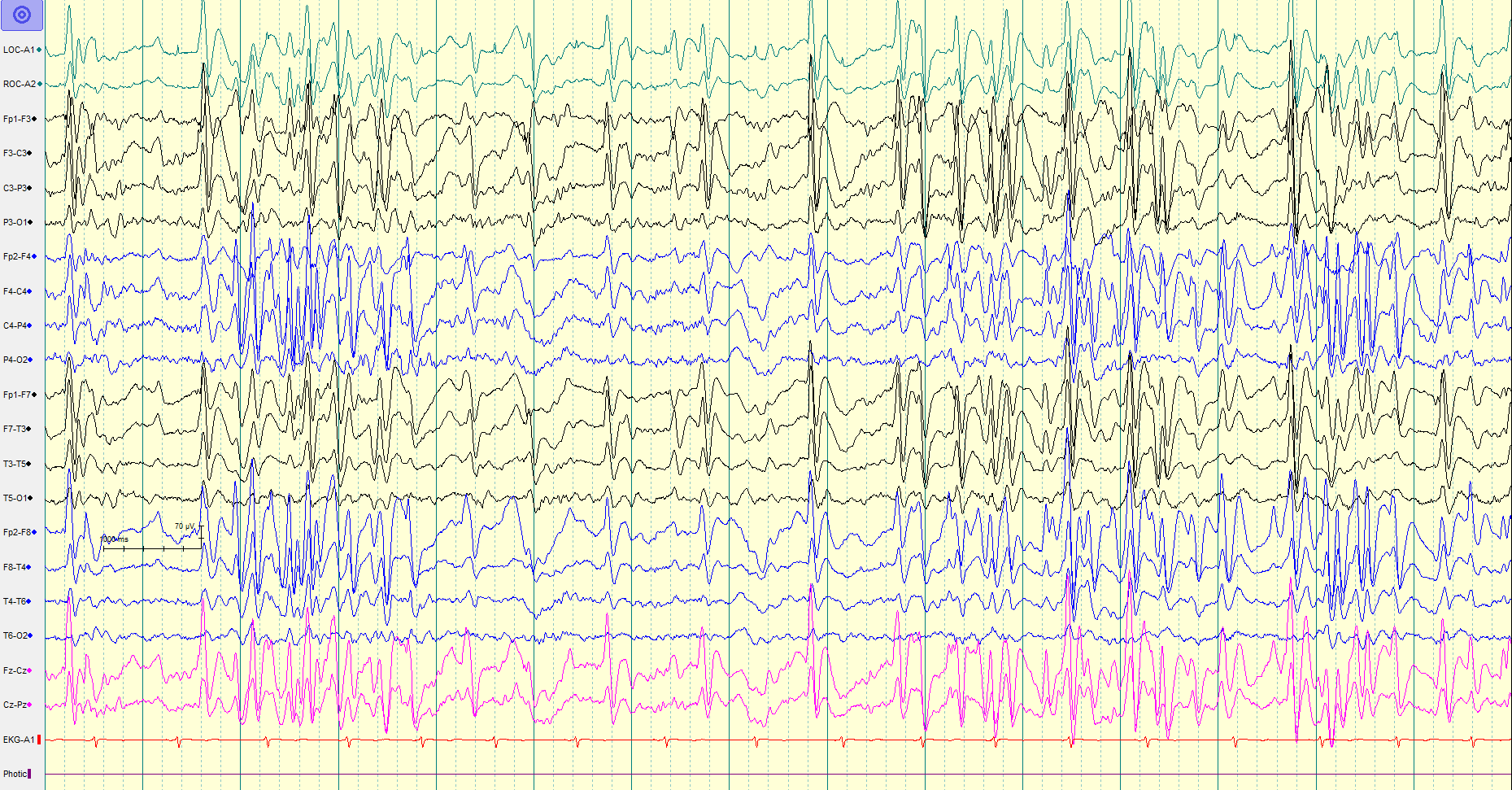
Electroencephalogram in Lennox-Gastaut Syndrome. This image represents a typical EEG pattern in a patient with Lennox-Gastaut Syndrome, demonstrating paroxysmal generalized fast activity in the background of generalized slow (1.5-2 Hz) spike-and-wave activity.
C Amrutkar, MD, and RM Riel-Romero, MD.
References
Markand ON. Lennox-Gastaut syndrome (childhood epileptic encephalopathy). Journal of clinical neurophysiology : official publication of the American Electroencephalographic Society. 2003 Nov-Dec:20(6):426-41 [PubMed PMID: 14734932]
Archer JS, Warren AE, Jackson GD, Abbott DF. Conceptualizing lennox-gastaut syndrome as a secondary network epilepsy. Frontiers in neurology. 2014:5():225. doi: 10.3389/fneur.2014.00225. Epub 2014 Oct 30 [PubMed PMID: 25400619]
Camfield PR. Definition and natural history of Lennox-Gastaut syndrome. Epilepsia. 2011 Aug:52 Suppl 5():3-9. doi: 10.1111/j.1528-1167.2011.03177.x. Epub [PubMed PMID: 21790560]
Khan S, Al Baradie R. Epileptic encephalopathies: an overview. Epilepsy research and treatment. 2012:2012():403592. doi: 10.1155/2012/403592. Epub 2012 Nov 20 [PubMed PMID: 23213494]
Level 3 (low-level) evidenceCross JH, Benítez A, Roth J, Andrews JS, Shah D, Butcher E, Jones A, Sullivan J. A comprehensive systematic literature review of the burden of illness of Lennox-Gastaut syndrome on patients, caregivers, and society. Epilepsia. 2024 May:65(5):1224-1239. doi: 10.1111/epi.17932. Epub 2024 Mar 8 [PubMed PMID: 38456647]
Level 1 (high-level) evidenceStrzelczyk A, Zuberi SM, Striano P, Rosenow F, Schubert-Bast S. The burden of illness in Lennox-Gastaut syndrome: a systematic literature review. Orphanet journal of rare diseases. 2023 Mar 1:18(1):42. doi: 10.1186/s13023-023-02626-4. Epub 2023 Mar 1 [PubMed PMID: 36859290]
Level 1 (high-level) evidenceTrevathan E, Murphy CC, Yeargin-Allsopp M. Prevalence and descriptive epidemiology of Lennox-Gastaut syndrome among Atlanta children. Epilepsia. 1997 Dec:38(12):1283-8 [PubMed PMID: 9578523]
Level 2 (mid-level) evidenceOhtahara S, Yamatogi Y, Ohtsuka Y. Prognosis of the Lennox syndrome-long-term clinical and electroencephalographic follow-up study, especially with special reference to relationship with the West syndrome. Folia psychiatrica et neurologica japonica. 1976:30(3):275-87 [PubMed PMID: 992512]
Widdess-Walsh P, Dlugos D, Fahlstrom R, Joshi S, Shellhaas R, Boro A, Sullivan J, Geller E, EPGP Investigators. Lennox-Gastaut syndrome of unknown cause: phenotypic characteristics of patients in the Epilepsy Phenome/Genome Project. Epilepsia. 2013 Nov:54(11):1898-904. doi: 10.1111/epi.12395. Epub 2013 Oct 7 [PubMed PMID: 24116958]
Selmer KK, Lund C, Brandal K, Undlien DE, Brodtkorb E. SCN1A mutation screening in adult patients with Lennox-Gastaut syndrome features. Epilepsy & behavior : E&B. 2009 Nov:16(3):555-7. doi: 10.1016/j.yebeh.2009.08.021. Epub 2009 Sep 24 [PubMed PMID: 19782004]
Epi4K Consortium, Epilepsy Phenome/Genome Project, Allen AS, Berkovic SF, Cossette P, Delanty N, Dlugos D, Eichler EE, Epstein MP, Glauser T, Goldstein DB, Han Y, Heinzen EL, Hitomi Y, Howell KB, Johnson MR, Kuzniecky R, Lowenstein DH, Lu YF, Madou MR, Marson AG, Mefford HC, Esmaeeli Nieh S, O'Brien TJ, Ottman R, Petrovski S, Poduri A, Ruzzo EK, Scheffer IE, Sherr EH, Yuskaitis CJ, Abou-Khalil B, Alldredge BK, Bautista JF, Berkovic SF, Boro A, Cascino GD, Consalvo D, Crumrine P, Devinsky O, Dlugos D, Epstein MP, Fiol M, Fountain NB, French J, Friedman D, Geller EB, Glauser T, Glynn S, Haut SR, Hayward J, Helmers SL, Joshi S, Kanner A, Kirsch HE, Knowlton RC, Kossoff EH, Kuperman R, Kuzniecky R, Lowenstein DH, McGuire SM, Motika PV, Novotny EJ, Ottman R, Paolicchi JM, Parent JM, Park K, Poduri A, Scheffer IE, Shellhaas RA, Sherr EH, Shih JJ, Singh R, Sirven J, Smith MC, Sullivan J, Lin Thio L, Venkat A, Vining EP, Von Allmen GK, Weisenberg JL, Widdess-Walsh P, Winawer MR. De novo mutations in epileptic encephalopathies. Nature. 2013 Sep 12:501(7466):217-21. doi: 10.1038/nature12439. Epub 2013 Aug 11 [PubMed PMID: 23934111]
Level 2 (mid-level) evidenceLund C, Brodtkorb E, Øye AM, Røsby O, Selmer KK. CHD2 mutations in Lennox-Gastaut syndrome. Epilepsy & behavior : E&B. 2014 Apr:33():18-21. doi: 10.1016/j.yebeh.2014.02.005. Epub 2014 Mar 12 [PubMed PMID: 24614520]
Capelli LP, Krepischi AC, Gurgel-Giannetti J, Mendes MF, Rodrigues T, Varela MC, Koiffmann CP, Rosenberg C. Deletion of the RMGA and CHD2 genes in a child with epilepsy and mental deficiency. European journal of medical genetics. 2012 Feb:55(2):132-4. doi: 10.1016/j.ejmg.2011.10.004. Epub 2011 Nov 25 [PubMed PMID: 22178256]
Level 3 (low-level) evidenceSullivan J, Benítez A, Roth J, Andrews JS, Shah D, Butcher E, Jones A, Cross JH. A systematic literature review on the global epidemiology of Dravet syndrome and Lennox-Gastaut syndrome: Prevalence, incidence, diagnosis, and mortality. Epilepsia. 2024 May:65(5):1240-1263. doi: 10.1111/epi.17866. Epub 2024 Jan 22 [PubMed PMID: 38252068]
Level 1 (high-level) evidenceBourgeois BF, Douglass LM, Sankar R. Lennox-Gastaut syndrome: a consensus approach to differential diagnosis. Epilepsia. 2014 Sep:55 Suppl 4():4-9. doi: 10.1111/epi.12567. Epub [PubMed PMID: 25284032]
Level 3 (low-level) evidenceHeiskala H. Community-based study of Lennox-Gastaut syndrome. Epilepsia. 1997 May:38(5):526-31 [PubMed PMID: 9184597]
Level 2 (mid-level) evidenceAsadi-Pooya AA, Sharifzade M. Lennox-Gastaut syndrome in south Iran: electro-clinical manifestations. Seizure. 2012 Dec:21(10):760-3. doi: 10.1016/j.seizure.2012.08.003. Epub 2012 Aug 22 [PubMed PMID: 22921514]
Level 2 (mid-level) evidenceCrumrine PK. Lennox-Gastaut syndrome. Journal of child neurology. 2002 Jan:17 Suppl 1():S70-5 [PubMed PMID: 11918467]
Goldsmith IL, Zupanc ML, Buchhalter JR. Long-term seizure outcome in 74 patients with Lennox-Gastaut syndrome: effects of incorporating MRI head imaging in defining the cryptogenic subgroup. Epilepsia. 2000 Apr:41(4):395-9 [PubMed PMID: 10756403]
Level 2 (mid-level) evidenceAsadi-Pooya AA. Lennox-Gastaut syndrome: a comprehensive review. Neurological sciences : official journal of the Italian Neurological Society and of the Italian Society of Clinical Neurophysiology. 2018 Mar:39(3):403-414. doi: 10.1007/s10072-017-3188-y. Epub 2017 Nov 9 [PubMed PMID: 29124439]
Poulen G, Gélisse P, Crespel A, Chan-Seng E, Moser PO, Coubes P. Does deep brain stimulation of the anterior nucleus of the thalamus represent the future of Lennox-Gastaut syndrome? Journal of neurology. 2025 Apr 3:272(4):312. doi: 10.1007/s00415-025-13053-9. Epub 2025 Apr 3 [PubMed PMID: 40180621]
Pillay N, Archer JS, Badawy RA, Flanagan DF, Berkovic SF, Jackson G. Networks underlying paroxysmal fast activity and slow spike and wave in Lennox-Gastaut syndrome. Neurology. 2013 Aug 13:81(7):665-73. doi: 10.1212/WNL.0b013e3182a08f6a. Epub 2013 Jul 17 [PubMed PMID: 23864316]
Arzimanoglou A, French J, Blume WT, Cross JH, Ernst JP, Feucht M, Genton P, Guerrini R, Kluger G, Pellock JM, Perucca E, Wheless JW. Lennox-Gastaut syndrome: a consensus approach on diagnosis, assessment, management, and trial methodology. The Lancet. Neurology. 2009 Jan:8(1):82-93. doi: 10.1016/S1474-4422(08)70292-8. Epub [PubMed PMID: 19081517]
Level 3 (low-level) evidenceBare MA, Glauser TA, Strawsburg RH. Need for electroencephalogram video confirmation of atypical absence seizures in children with Lennox-Gastaut syndrome. Journal of child neurology. 1998 Oct:13(10):498-500 [PubMed PMID: 9796756]
Level 2 (mid-level) evidenceGlauser TA. Following catastrophic epilepsy patients from childhood to adulthood. Epilepsia. 2004:45 Suppl 5():23-6 [PubMed PMID: 15283708]
Specchio N, Wirrell EC, Scheffer IE, Nabbout R, Riney K, Samia P, Guerreiro M, Gwer S, Zuberi SM, Wilmshurst JM, Yozawitz E, Pressler R, Hirsch E, Wiebe S, Cross HJ, Perucca E, Moshé SL, Tinuper P, Auvin S. International League Against Epilepsy classification and definition of epilepsy syndromes with onset in childhood: Position paper by the ILAE Task Force on Nosology and Definitions. Epilepsia. 2022 Jun:63(6):1398-1442. doi: 10.1111/epi.17241. Epub 2022 May 3 [PubMed PMID: 35503717]
Oguni H, Hayashi K, Osawa M. Long-term prognosis of Lennox-Gastaut syndrome. Epilepsia. 1996:37 Suppl 3():44-7 [PubMed PMID: 8681912]
Asadi-Pooya AA, Dlugos D, Skidmore C, Sperling MR. Atlas of Electroencephalography, 3rd Edition. Epileptic disorders : international epilepsy journal with videotape. 2017 Sep 1:19(3):384. doi: 10.1684/epd.2017.0934. Epub [PubMed PMID: 28872032]
Warren AEL, Patel AD, Helen Cross J, Clarke DF, Dalic LJ, Grinspan ZM, Conecker G, Knowles JK. Mobilizing a New Era in Lennox-Gastaut Syndrome Treatment and Prevention. Epilepsy currents. 2025 Mar 25:():15357597251321926. doi: 10.1177/15357597251321926. Epub 2025 Mar 25 [PubMed PMID: 40161505]
Shyu HY, Lin JH, Chen C, Kwan SY, Yiu CH. An atypical case of Lennox-Gastaut syndrome not associated with mental retardation: a nosological issue. Seizure. 2011 Dec:20(10):820-3. doi: 10.1016/j.seizure.2011.08.001. Epub 2011 Aug 21 [PubMed PMID: 21862354]
Level 3 (low-level) evidenceMichoulas A, Farrell K. Medical management of Lennox-Gastaut syndrome. CNS drugs. 2010 May:24(5):363-74. doi: 10.2165/11530220-000000000-00000. Epub [PubMed PMID: 20158289]
Samanta D. Management of Lennox-Gastaut syndrome beyond childhood: A comprehensive review. Epilepsy & behavior : E&B. 2021 Jan:114(Pt A):107612. doi: 10.1016/j.yebeh.2020.107612. Epub 2020 Nov 24 [PubMed PMID: 33243685]
Motte J, Trevathan E, Arvidsson JF, Barrera MN, Mullens EL, Manasco P. Lamotrigine for generalized seizures associated with the Lennox-Gastaut syndrome. Lamictal Lennox-Gastaut Study Group. The New England journal of medicine. 1997 Dec 18:337(25):1807-12 [PubMed PMID: 9400037]
Level 1 (high-level) evidenceSachdeo RC, Glauser TA, Ritter F, Reife R, Lim P, Pledger G. A double-blind, randomized trial of topiramate in Lennox-Gastaut syndrome. Topiramate YL Study Group. Neurology. 1999 Jun 10:52(9):1882-7 [PubMed PMID: 10371538]
Level 1 (high-level) evidenceGlauser T, Kluger G, Sachdeo R, Krauss G, Perdomo C, Arroyo S. Rufinamide for generalized seizures associated with Lennox-Gastaut syndrome. Neurology. 2008 May 20:70(21):1950-8. doi: 10.1212/01.wnl.0000303813.95800.0d. Epub 2008 Apr 9 [PubMed PMID: 18401024]
Level 1 (high-level) evidenceNg YT, Conry J, Mitchell WG, Buchhalter J, Isojarvi J, Lee D, Drummond R, Chung S. Clobazam is equally safe and efficacious for seizures associated with Lennox-Gastaut syndrome across different age groups: Post hoc analyses of short- and long-term clinical trial results. Epilepsy & behavior : E&B. 2015 May:46():221-6. doi: 10.1016/j.yebeh.2015.01.037. Epub 2015 May 1 [PubMed PMID: 25940107]
Wheless JW, Isojarvi J, Lee D, Drummond R, Benbadis SR. Clobazam is efficacious for patients across the spectrum of disease severity of Lennox-Gastaut syndrome: post hoc analyses of clinical trial results by baseline seizure-frequency quartiles and VNS experience. Epilepsy & behavior : E&B. 2014 Dec:41():47-52. doi: 10.1016/j.yebeh.2014.09.019. Epub 2014 Oct 2 [PubMed PMID: 25282105]
Felbamate Study Group in Lennox-Gastaut Syndrome. Efficacy of felbamate in childhood epileptic encephalopathy (Lennox-Gastaut syndrome). The New England journal of medicine. 1993 Jan 7:328(1):29-33 [PubMed PMID: 8347179]
Level 1 (high-level) evidenceDevinsky O, Faught RE, Wilder BJ, Kanner AM, Kamin M, Kramer LD, Rosenberg A. Efficacy of felbamate monotherapy in patients undergoing presurgical evaluation of partial seizures. Epilepsy research. 1995 Mar:20(3):241-6 [PubMed PMID: 7796796]
Level 1 (high-level) evidenceO'Neil MG, Perdun CS, Wilson MB, McGown ST, Patel S. Felbamate-associated fatal acute hepatic necrosis. Neurology. 1996 May:46(5):1457-9 [PubMed PMID: 8628501]
Level 3 (low-level) evidenceJaraba S, Santamarina E, Miró J, Toledo M, Molins A, Burcet J, Becerra JL, Raspall M, Pico G, Miravet E, Cano A, Fossas P, Fernández S, Falip M. Rufinamide in children and adults in routine clinical practice. Acta neurologica Scandinavica. 2017 Jan:135(1):122-128. doi: 10.1111/ane.12572. Epub 2016 Feb 29 [PubMed PMID: 26923380]
Devinsky O, Marsh E, Friedman D, Thiele E, Laux L, Sullivan J, Miller I, Flamini R, Wilfong A, Filloux F, Wong M, Tilton N, Bruno P, Bluvstein J, Hedlund J, Kamens R, Maclean J, Nangia S, Singhal NS, Wilson CA, Patel A, Cilio MR. Cannabidiol in patients with treatment-resistant epilepsy: an open-label interventional trial. The Lancet. Neurology. 2016 Mar:15(3):270-8. doi: 10.1016/S1474-4422(15)00379-8. Epub 2015 Dec 24 [PubMed PMID: 26724101]
Free T, Grossoehme DH, Richner G, Brown MF, Friebert S. Characterizing the Population of a Medical Cannabis Clinic in a Pediatric Hospital. Journal of palliative medicine. 2025 Apr 3:():. doi: 10.1089/jpm.2024.0533. Epub 2025 Apr 3 [PubMed PMID: 40180570]
Cerulli Irelli E, Mazzeo A, Caraballo RH, Perulli M, Moloney PB, Peña-Ceballos J, Rubino M, Mieszczanek KM, Santangelo A, Licchetta L, De Giorgis V, Reyes Valenzuela G, Casellato S, Cesaroni E, Operto FF, Domínguez-Carral J, Ramírez-Camacho A, Ferretti A, Santangelo G, Aledo-Serrano A, Rüegger A, Mancardi MM, Prato G, Riva A, Bergonzini L, Cordelli DM, Bonanni P, Bisulli F, Di Gennaro G, Matricardi S, Striano P, Delanty N, Marini C, Battaglia D, Di Bonaventura C, Ramantani G, Gardella E, GENE‐CBD Study Group, Orsini A, Coppola A. Expanding the therapeutic role of highly purified cannabidiol in monogenic epilepsies: A multicenter real-world study. Epilepsia. 2025 Mar 24:():. doi: 10.1111/epi.18378. Epub 2025 Mar 24 [PubMed PMID: 40126049]
Level 3 (low-level) evidenceAsadi-Pooya AA, Emami M, Ashjazadeh N, Nikseresht A, Shariat A, Petramfar P, Yousefipour G, Borhani-Haghighi A, Izadi S, Rahimi-Jaberi A. Reasons for uncontrolled seizures in adults; the impact of pseudointractability. Seizure. 2013 May:22(4):271-4. doi: 10.1016/j.seizure.2013.01.010. Epub 2013 Feb 1 [PubMed PMID: 23375939]
Level 2 (mid-level) evidencePera MC, Randazzo G, Masnada S, Dontin SD, De Giorgis V, Balottin U, Veggiotti P. Intravenous methylprednisolone pulse therapy for children with epileptic encephalopathy. Functional neurology. 2015 Jul-Sep:30(3):173-9 [PubMed PMID: 26910177]
Mikati MA, Kurdi R, El-Khoury Z, Rahi A, Raad W. Intravenous immunoglobulin therapy in intractable childhood epilepsy: open-label study and review of the literature. Epilepsy & behavior : E&B. 2010 Jan:17(1):90-4. doi: 10.1016/j.yebeh.2009.10.020. Epub 2009 Dec 9 [PubMed PMID: 20004620]
Caraballo RH, Fortini S, Fresler S, Armeno M, Ariela A, Cresta A, Mestre G, Escobal N. Ketogenic diet in patients with Lennox-Gastaut syndrome. Seizure. 2014 Oct:23(9):751-5. doi: 10.1016/j.seizure.2014.06.005. Epub 2014 Jun 19 [PubMed PMID: 25011392]
Level 2 (mid-level) evidenceSharma S, Jain P, Gulati S, Sankhyan N, Agarwala A. Use of the modified Atkins diet in Lennox Gastaut syndrome. Journal of child neurology. 2015 Apr:30(5):576-9. doi: 10.1177/0883073814527162. Epub 2014 Mar 20 [PubMed PMID: 24659735]
Level 2 (mid-level) evidenceKim SH, Kang HC, Lee EJ, Lee JS, Kim HD. Low glycemic index treatment in patients with drug-resistant epilepsy. Brain & development. 2017 Sep:39(8):687-692. doi: 10.1016/j.braindev.2017.03.027. Epub 2017 Apr 18 [PubMed PMID: 28431772]
Lemmon ME, Terao NN, Ng YT, Reisig W, Rubenstein JE, Kossoff EH. Efficacy of the ketogenic diet in Lennox-Gastaut syndrome: a retrospective review of one institution's experience and summary of the literature. Developmental medicine and child neurology. 2012 May:54(5):464-8. doi: 10.1111/j.1469-8749.2012.04233.x. Epub 2012 Mar 22 [PubMed PMID: 22443637]
Level 2 (mid-level) evidenceDouglass LM, Salpekar J. Surgical options for patients with Lennox-Gastaut syndrome. Epilepsia. 2014 Sep:55 Suppl 4():21-8. doi: 10.1111/epi.12742. Epub [PubMed PMID: 25284034]
Asadi-Pooya AA, Malekmohamadi Z, Kamgarpour A, Rakei SM, Taghipour M, Ashjazadeh N, Inaloo S, Razmkon A, Zare Z. Corpus callosotomy is a valuable therapeutic option for patients with Lennox-Gastaut syndrome and medically refractory seizures. Epilepsy & behavior : E&B. 2013 Nov:29(2):285-8. doi: 10.1016/j.yebeh.2013.08.011. Epub 2013 Sep 5 [PubMed PMID: 24012506]
Asadi-Pooya AA, Sharan A, Nei M, Sperling MR. Corpus callosotomy. Epilepsy & behavior : E&B. 2008 Aug:13(2):271-8. doi: 10.1016/j.yebeh.2008.04.020. Epub 2008 Jun 6 [PubMed PMID: 18539083]
Kerr M, Kluger G, Philip S. Evolution and management of Lennox-Gastaut syndrome through adolescence and into adulthood: are seizures always the primary issue? Epileptic disorders : international epilepsy journal with videotape. 2011 May:13 Suppl 1():S15-26. doi: 10.1684/epd.2011.0409. Epub [PubMed PMID: 21669559]
Wirrell E, Farrell K, Whiting S. The epileptic encephalopathies of infancy and childhood. The Canadian journal of neurological sciences. Le journal canadien des sciences neurologiques. 2005 Nov:32(4):409-18 [PubMed PMID: 16408569]
Zuberi SM, Wirrell E, Yozawitz E, Wilmshurst JM, Specchio N, Riney K, Pressler R, Auvin S, Samia P, Hirsch E, Galicchio S, Triki C, Snead OC, Wiebe S, Cross JH, Tinuper P, Scheffer IE, Perucca E, Moshé SL, Nabbout R. ILAE classification and definition of epilepsy syndromes with onset in neonates and infants: Position statement by the ILAE Task Force on Nosology and Definitions. Epilepsia. 2022 Jun:63(6):1349-1397. doi: 10.1111/epi.17239. Epub 2022 May 3 [PubMed PMID: 35503712]
Doose H. Myoclonic-astatic epilepsy. Epilepsy research. Supplement. 1992:6():163-8 [PubMed PMID: 1418479]
Cross JH, Auvin S, Falip M, Striano P, Arzimanoglou A. Expert Opinion on the Management of Lennox-Gastaut Syndrome: Treatment Algorithms and Practical Considerations. Frontiers in neurology. 2017:8():505. doi: 10.3389/fneur.2017.00505. Epub 2017 Sep 29 [PubMed PMID: 29085326]
Level 3 (low-level) evidencePeron A, Catusi I, Recalcati MP, Calzari L, Larizza L, Vignoli A, Canevini MP. Ring Chromosome 20 Syndrome: Genetics, Clinical Characteristics, and Overlapping Phenotypes. Frontiers in neurology. 2020:11():613035. doi: 10.3389/fneur.2020.613035. Epub 2020 Dec 8 [PubMed PMID: 33363513]
Pampiglione G, Harden A. Neurophysiological identification of a late infantile form of 'neuronal lipidosis'. Journal of neurology, neurosurgery, and psychiatry. 1973 Feb:36(1):68-74 [PubMed PMID: 4691693]
Vignoli A, Oggioni G, De Maria G, Peron A, Savini MN, Zambrelli E, Chiesa V, La Briola F, Turner K, Canevini MP. Lennox-Gastaut syndrome in adulthood: Long-term clinical follow-up of 38 patients and analysis of their recorded seizures. Epilepsy & behavior : E&B. 2017 Dec:77():73-78. doi: 10.1016/j.yebeh.2017.09.006. Epub 2017 Nov 7 [PubMed PMID: 29126048]
Berg AT, Nickels K, Wirrell EC, Geerts AT, Callenbach PM, Arts WF, Rios C, Camfield PR, Camfield CS. Mortality risks in new-onset childhood epilepsy. Pediatrics. 2013 Jul:132(1):124-31. doi: 10.1542/peds.2012-3998. Epub 2013 Jun 10 [PubMed PMID: 23753097]
Level 2 (mid-level) evidenceWang XQ, Lv B, Wang HF, Zhang X, Yu SY, Huang XS, Zhang JT, Tian CL, Lang SY. Lamotrigine induced DIHS/DRESS: Manifestations, treatment, and outcome in 57 patients. Clinical neurology and neurosurgery. 2015 Nov:138():1-7. doi: 10.1016/j.clineuro.2015.07.008. Epub 2015 Jul 8 [PubMed PMID: 26209753]
Ignaszewski M, Ignaszewski MJ, Kohlitz P. Lamotrigine-Associated Hemophagocytic Lymphohistiocytosis. American journal of therapeutics. 2017 Jul/Aug:24(4):e493. doi: 10.1097/MJT.0000000000000515. Epub [PubMed PMID: 28005556]