Introduction
Retinoblastoma is an uncommon malignancy, occurring in 1 per 18,000 births, but it is the most common primary intraocular cancer of childhood, accounting for 3% of pediatric malignancies.[1][2] This tumor is also the 2nd most prevalent intraocular malignancy after uveal melanoma. In specialized care centers, survival rates reach 95%, with vision preserved in most cases, though outcomes are poorer in developing countries.
Histologically, retinoblastoma consists of basophilic cells with hyperchromatic nuclei and scant cytoplasm. Most tumors are undifferentiated, but some show differentiation by forming structures known as rosettes. Growth patterns include endophytic (vitreous seeding), exophytic (subretinal seeding), and mixed. Tumor invasion can extend into the optic nerve, subarachnoid space, and brain, and metastases can occur in the regional lymph nodes, liver, lungs, bones, and brain.[3]
This aggressive neoplasm originates in the retina and primarily affects children younger than 5 years. With an incidence of 1 in 15,000 to 20,000 live births, retinoblastoma remains a significant contributor to global childhood cancer mortality. Advances in early diagnosis, imaging, and treatment have improved survival rates, particularly in high-resource settings. However, disparities persist due to delays in diagnosis, limited access to specialized care, and socioeconomic factors.
Retinoblastoma arises from a mutation in the RB1 tumor suppressor gene on chromosome 13q14, which regulates cell cycle progression. The inactivation of this gene leads to uncontrolled retinal cell proliferation. Approximately 45% of cases are hereditary, which is often bilateral, while 55% are nonhereditary and typically unilateral. Understanding the genetic basis of retinoblastoma is crucial for risk assessment, genetic counseling, and family surveillance. Common presentations include leukocoria (white pupillary reflex), strabismus, and, less commonly, red eye or decreased visual acuity.[4]
Parents or caregivers typically notice early signs, prompting evaluation by an ophthalmologist. Advanced cases may present with proptosis, orbital inflammation, or secondary glaucoma due to tumor extension beyond the globe. While leukocoria is the hallmark feature, educating caregivers and primary care providers about subtle warning signs is crucial, as delays in diagnosis are associated with poorer outcomes.
Retinoblastoma diagnosis relies on clinical examination, imaging, and molecular testing. Ophthalmic evaluation under anesthesia remains the gold standard, allowing detailed retinal assessment with indirect ophthalmoscopy. Imaging modalities such as ultrasound, magnetic resonance imaging (MRI), and optical coherence tomography (OCT) help confirm the diagnosis, evaluate intraocular disease extent, and rule out extraocular or metastatic spread. MRI is particularly useful for detecting optic nerve invasion, a key prognostic factor.
Molecular analysis of the RB1 gene confirms hereditary retinoblastoma and guides family counseling. Identifying germline mutations allows targeted screening of at-risk relatives, facilitating early detection. Emerging liquid biopsy techniques, such as tumor-derived DNA detection in aqueous humor or plasma, offer a promising noninvasive diagnostic approach.[5]
Accurate staging and classification are essential for determining treatment strategies and predicting outcomes. Retinoblastoma is classified using the International Intraocular Retinoblastoma Classification (IIRC) for intraocular tumors and the Tumor, Node, Metastasis (TNM) system for extraocular disease. The IIRC categorizes retinoblastoma into 5 groups (A–E) based on tumor size, location, and extent. Group A represents the least severe cases, and group E comprises advanced disease requiring enucleation. The TNM system accounts for systemic spread, including lymph node involvement and distant metastases.
Management has evolved significantly, with a growing emphasis on organ preservation, vision salvage, and minimizing long-term treatment complications. Treatment decisions depend on the extent of the disease, the patient's age, and access to specialized resources.[6]
Focal therapies such as cryotherapy, thermotherapy, and laser photocoagulation are commonly used for small, localized tumors (groups A and B). These modalities provide excellent tumor control with minimal systemic toxicity, making them the preferred treatment for early-stage disease.
For larger tumors (groups C and D), systemic chemotherapy is often used to shrink the neoplasm before applying focal treatments, a strategy known as chemoreduction. This approach has significantly reduced the need for enucleation and external beam radiotherapy (EBRT), preserving the eye and minimizing long-term complications such as secondary malignancies.
Intraarterial chemotherapy (IAC) has revolutionized retinoblastoma treatment by delivering chemotherapy directly into the ophthalmic artery. This method achieves high local drug concentrations while limiting systemic toxicity, making it particularly effective for advanced intraocular disease (groups C–E) and improving organ preservation rates.
For tumors with vitreous seeding, intravitreal chemotherapy (IVC) provides targeted drug delivery into the vitreous cavity. Once considered controversial due to concerns about extraocular spread, advancements in delivery techniques have made IVC a safe and effective option for controlling vitreous disease.[7]
Enucleation remains the primary treatment for advanced cases (group E or extraocular retinoblastoma) where vision preservation is not possible. Orbital exenteration may be necessary in cases of extensive orbital involvement. Postoperative adjuvant therapy, including chemotherapy or radiotherapy, is often required to address residual disease and reduce the risk of metastasis.
Emerging targeted therapies, such as small-molecule inhibitors, gene therapy, and immunotherapy, are under investigation for refractory or metastatic retinoblastoma. While still experimental, these approaches have the potential to improve outcomes for high-risk patients.
Managing retinoblastoma requires an interprofessional team, including ophthalmologists, pediatric oncologists, genetic counselors, radiologists, and specialized nursing staff. Genetic counseling is essential, particularly for families with hereditary retinoblastoma, as it provides critical information on recurrence risk, screening strategies, and reproductive options.[8]
Nurses play a critical role in patient education, symptom management, and psychosocial support, ensuring comprehensive care for children with retinoblastoma and their families. However, timely diagnosis and treatment remain significant challenges in resource-limited settings. Strengthening the healthcare infrastructure, training personnel, and implementing awareness programs are essential to reducing disparities and improving global outcomes.
Retinoblastoma requires early diagnosis and prompt intervention to maximize survival and preserve vision. Advances in diagnostic techniques, molecular testing, and therapeutic approaches have transformed care, emphasizing eye preservation and minimizing treatment-related morbidity. Ongoing research and interprofessional collaboration remain vital to addressing existing gaps and ensuring equitable access to state-of-the-art care for all patients, regardless of geographic or socioeconomic barriers.[9] This introduction sets the stage for exploring the intricacies of retinoblastoma management and the evolving landscape of pediatric ocular oncology.
Etiology
Register For Free And Read The Full Article
 Search engine and full access to all medical articles
Search engine and full access to all medical articles- 10 free questions in your specialty
- Free CME/CE Activities
- Free daily question in your email
- Save favorite articles to your dashboard
- Emails offering discounts
Learn more about a Subscription to StatPearls Point-of-Care
Etiology
Genetic Basis of Retinoblastoma
Retinoblastoma is primarily caused by mutations in the RB1 tumor suppressor gene located on chromosome 13q14. The RB1 gene encodes the retinoblastoma protein (pRB), a key regulator of the cell cycle.[10] Under normal conditions, pRB prevents uncontrolled cell proliferation by inhibiting the transition from the G1 phase to the S phase. When both alleles of RB1 are mutated or inactivated, pRB loses its regulatory function, leading to unchecked retinal cell growth and tumor formation.
In bilateral retinoblastoma, the mutation is germline in 98% of cases, although only 5% of cases have a family history. Approximately 95% of retinoblastoma cases occur sporadically, with 60% presenting as unilateral disease without an associated germline mutation. The remaining cases involve germline mutations, often resulting in multiple tumor development.[11]
The development of retinoblastoma follows Alfred Knudson’s 2-hit hypothesis. In hereditary cases, the 1st "hit" occurs when an individual inherits a defective RB1 allele as a germline mutation in all body cells. The 2nd "hit" is a somatic mutation that inactivates the remaining functional RB1 allele within retinal cells, triggering tumor formation. In nonhereditary cases, both mutations arise sporadically within retinal cells, leading to tumorigenesis.[12]
Hereditary Retinoblastoma
Hereditary cases account for approximately 45% of all retinoblastomas. These cases often present bilaterally or multifocally but within the same eye. Patients with a germline mutation in the RB1 gene face an increased risk of retinoblastoma and secondary malignancies, including osteosarcoma, soft tissue sarcomas, and melanoma.
Familial retinoblastoma follows an autosomal dominant inheritance pattern with high penetrance. A mutation in one RB1 allele is present in all body cells, and malignant transformation occurs when a 2nd mutation affects the remaining allele. Since the mutation exists in every cell, many affected children develop bilateral and multifocal tumors.
Individuals with hereditary retinoblastoma face a significant risk of nonocular cancers, such as pineoblastoma, osteosarcoma, soft tissue sarcomas, and melanomas, which tend to occur within specific age groups.[13] The likelihood of developing a secondary malignancy is 6%, but the risk increases fivefold when EBRT is used to treat the primary tumor.
Sporadic (Nonhereditary) Retinoblastoma
Sporadic cases account for approximately 55% of all retinoblastomas. These cases are typically unilateral and unifocal, meaning a single tumor develops in 1 eye. Unlike hereditary forms, both RB1 mutations occur sporadically within retinal cells without an underlying germline mutation. Since the mutation is not present in all body cells, patients are less likely to develop secondary malignancies.
Nonheritable retinoblastomas are not transmitted to offspring. In patients with unilateral retinoblastoma but without a significant family history, the condition is classified as nonheritable, and the risk of occurrence in siblings or the patient’s children is only 1%. Nearly 90% of unilateral retinoblastoma cases fall under this category.
Retinoblastoma is a malignant tumor of the retina that primarily affects children younger than 5. The lesion's development is strongly linked to genetic mutations that disrupt cellular growth and apoptosis regulation, leading to uncontrolled retinal cell proliferation. Understanding these genetic and molecular mechanisms provides critical insight into retinoblastoma pathogenesis and informs targeted approaches to diagnosis and treatment.[14]
RB1-Independent Pathways
Although most retinoblastoma cases result from RB1 mutations, approximately 2% develop through RB1-independent mechanisms. Some nonhereditary cases involve amplification of the MYCN oncogene, which drives rapid cell proliferation and tumor growth. MYCN-amplified retinoblastomas tend to be aggressive, presenting as unilateral tumors in very young infants.
In some cases, RB1 is not inactivated by genetic mutations but by epigenetic silencing, such as promoter hypermethylation. This modification reduces RB1 expression, leading to tumor formation.[15]
Molecular Pathways in Retinoblastoma
The molecular mechanisms underlying retinoblastoma involve several disrupted pathways that contribute to tumor progression. Loss of RB1 function results in the uncontrolled activity of the E2F transcription factor, which drives the expression of genes involved in DNA synthesis and cell cycle progression. This unchecked activity leads to the proliferation of immature retinal cells.[16] Furthermore, the absence of functional RB1 impairs chromosomal stability, accumulating additional genetic alterations that contribute to tumor progression.[17]
Retinoblastoma tumors also exhibit increased angiogenesis, driven by factors like vascular endothelial growth factor (VEGF), which supports tumor growth and sustains the metabolic needs of rapidly dividing cancer cells.[18] Additionally, retinoblastoma cells acquire the ability to evade programmed cell death (apoptosis), often through mechanisms such as overexpression of antiapoptotic proteins like the Bcl-2 family and activation of survival pathways, such as the PI3K/Akt signaling pathway. These molecular alterations further facilitate the growth and persistence of the tumor.[19]
Environmental and Nongenetic Factors
While genetic mutations primarily drive retinoblastoma development, certain nongenetic factors may influence disease risk. Advanced paternal age has been weakly associated with an increased likelihood of germline RB1 mutations, possibly due to accumulated DNA replication errors in sperm.[20]
Environmental mutagens, including radiation, chemical exposures, and other carcinogens encountered during pregnancy, have been suggested as potential contributors to sporadic mutations in fetal retinal cells. However, direct evidence linking these exposures to retinoblastoma remains limited.[21]
Maternal health may also contribute to disease risk. Conditions such as diabetes and gestational infections could create an intrauterine environment that increases the susceptibility of developing retinal cells to genetic damage.[22]
Genetic Counseling Implications
Children with hereditary retinoblastoma have a 50% chance of passing the germline RB1 mutation to their offspring, making genetic counseling essential for affected families. Identifying mutation carriers among siblings and other relatives allows for early surveillance and timely intervention, reducing the risk of late-stage diagnosis. Genetic testing provides critical information to guide screening strategies and reproductive decisions.[23]
For newborns with a family history of retinoblastoma, regular retinal examinations should begin at birth to detect tumors early. Advances in molecular testing, including liquid biopsies that analyze tumor-derived DNA in blood or aqueous humor, have significantly improved early diagnosis and disease monitoring. These innovations enhance the ability to detect retinoblastoma before clinical symptoms appear, allowing for prompt treatment and better visual outcomes.
Table 1. Summary of Etiologic Subtypes
|
Subtype |
Genetic Alteration |
Presentation |
Other Clinical Features |
|
Hereditary |
Germline RB1 mutation |
Bilateral or multifocal; early onset |
Family history of retinoblastoma |
|
Nonhereditary |
Sporadic RB1 mutations |
Unilateral; unifocal |
No family history |
|
MYCN-Amplified |
MYCN oncogene amplification |
Aggressive; unilateral |
Seen in very young infants |
|
Epigenetic |
Promoter hypermethylation of RB1 |
Resembling sporadic cases |
Rare mechanism |
The etiology of retinoblastoma is multifaceted, with RB1 mutations serving as the central driver in most cases. Understanding the genetic basis of this malignancy is essential for early detection, risk assessment, and management. Advances in molecular diagnostics and a deeper understanding of RB1-independent mechanisms hold promise for improving outcomes and guiding personalized treatment strategies for this potentially sight-threatening disease.[24]
Epidemiology
Incidence
Retinoblastoma is the most common primary intraocular malignancy in children, representing approximately 3% of all pediatric tumors. This pathology is the 2nd most common intraocular malignant tumor overall. The incidence ranges from 1 in 14,000 to 1 in 20,000 live births, with approximately 300 new cases diagnosed annually in the U.S.[25] While relatively rare, retinoblastoma’s potential for severe morbidity and mortality underscores the importance of understanding its epidemiological patterns.
The global incidence of retinoblastoma is estimated to be approximately 1 in 15,000 to 20,000 live births, resulting in around 8,000 new cases annually. This figure corresponds to about 10 to 15 cases per million children younger than 5. The incidence is relatively constant across populations, reflecting a stable mutation rate in the RB1 gene, which drives most cases.[26]
Retinoblastoma primarily affects children younger than 5, with a median age at diagnosis of 12 months for bilateral cases and 24 months for unilateral cases. Approximately 95% of cases are diagnosed before age 5, with 90% identified before age 3. Meanwhile, occurrence in older children and adults is exceedingly rare.
Incidence varies geographically, with reported rates of 6 cases per million in Mexico and 4 cases per million in the U.S. The highest incidence has been observed in India and Africa.[27] Survival rates and outcomes vary significantly between high-income (HICs) and low- and middle-income countries (LMICs). In HICs, survival rates exceed 95% due to early diagnosis, advanced medical infrastructure, and access to comprehensive care. Most cases are detected at an early stage, allowing for eye-preserving treatments. In LMICs, survival rates range from 30% to 60%, with even lower rates in some regions of sub-Saharan Africa and South Asia. Late diagnosis and limited access to specialized care often result in worse outcomes, frequently requiring enucleation or leading to extraocular spread and metastatic disease.
Retinoblastoma affects male and female individuals equally, showing no significant sex predilection. The disease also lacks ethnic or racial predisposition, with incidence rates remaining similar across different populations.
Hereditary vs. Sporadic Cases
Retinoblastoma is broadly categorized into hereditary and sporadic forms, each with distinct epidemiological patterns.[28] Hereditary retinoblastoma accounts for 45% of cases and often presents as bilateral or multifocal tumors. Affected individuals inherit a germline RB1 mutation, which carries a 50% chance of transmission to offspring. These cases tend to be diagnosed earlier than sporadic cases, with a median age of onset of 12 months. In addition to eye involvement, hereditary retinoblastoma is associated with an increased risk of secondary malignancies later in life, including osteosarcoma, soft tissue sarcoma, and melanoma.
Sporadic retinoblastoma comprises 55% of cases and typically manifests as a unilateral, unifocal tumor. Unlike hereditary cases, sporadic retinoblastoma results from 2 somatic mutations in the RB1 gene occurring within retinal cells. The median age of onset is later, around 24 months. Since these cases lack germline mutations, affected individuals do not have an elevated risk of developing secondary malignancies.
Survival Rates
Retinoblastoma is rarely fatal in HICs, with 5-year survival rates exceeding 95%. Early diagnosis and the use of eye-preserving treatments, such as IAC and focal therapies, contribute to these high survival rates. In contrast, survival rates in LMICs are significantly lower due to limiting factors, including late diagnosis, poor access to specialized care, and a high prevalence of advanced disease at the time of presentation. Many children in these regions present with extraocular retinoblastoma, a form of the disease that carries a poor prognosis and often results in fatality without aggressive treatment.[29]
Global Disparities
Global disparities exist in the management of retinoblastoma. In LMICs, delayed diagnosis often results from restrictive cultural beliefs, limited access to healthcare, and a lack of awareness among caregivers and primary care providers, contributing to late-stage presentations that significantly increase mortality rates. Furthermore, many LMICs lack the necessary infrastructure and trained personnel to provide advanced diagnostic imaging, chemotherapy, and radiotherapy, which are essential for effective treatment. Economic barriers also play a major role, as the high costs of treatment, including systemic chemotherapy and specialized surgeries like enucleation, create significant challenges in resource-limited settings.[30]
Genetic and Familial Patterns
Children with hereditary retinoblastoma have a 50% chance of passing the germline RB1 mutation to their offspring. Siblings of affected individuals also have an increased risk of developing retinoblastoma. Advances in genetic testing have made it possible to identify at-risk infants, enabling regular surveillance and early tumor detection, which significantly improves outcomes.[31]
Epidemiological Trends
Advances in early detection, imaging, and eye-preserving therapies have significantly improved survival rates in HICs, with a focus on minimizing treatment-related morbidity. The introduction of innovative treatments, such as IAC, IVC, and molecular targeted therapies, has revolutionized the management of retinoblastoma. Efforts to address global disparities in care are being made through organizations like the International Agency for the Prevention of Blindness (IAPB) and the Retinoblastoma World Alliance, which are improving awareness, training, and healthcare access in LMICs. Public health campaigns in LMICs aim to educate caregivers and healthcare providers about early signs, such as leukocoria, promoting earlier diagnosis.[32]
Table 2. Key Statistics for Retinoblastoma
| Global Incidence | 8,000 new cases annually |
| Survival Disparities |
HICs: >95% survival rate LMICs: 30-60% survival rate, with higher mortality rates in certain regions |
| Bilateral Cases | 25-30% of retinoblastoma cases; often associated with hereditary disease |
| Median Age at Diagnosis |
Bilateral cases: 12 months Unilateral cases: 24 months [33] |
The epidemiology of retinoblastoma reflects both the advancements in pediatric oncology and the persistent global disparities in healthcare access and outcomes. While survival rates in HICs are near 100%, children in low-resource settings face significant barriers to diagnosis and treatment, leading to high mortality rates. Efforts to bridge these gaps, including public health initiatives, improved genetic screening, and international collaborations, are essential to ensure that every child, regardless of geography or socioeconomic status, has access to life-saving care for retinoblastoma.
Pathophysiology
Oncogenesis
Retinoblastoma is a malignant neoplasm arising from immature retinal cells. As mentioned earlier, the evolution of this condition is triggered by mutations primarily in the RB1 tumor suppressor gene. These mutations lead to disruptions in cell cycle regulation, genomic stability, and programmed cell death, which collectively result in uncontrolled retinal cell proliferation and tumor formation.
The RB1 gene, located on chromosome 13q14, encodes pRB, a crucial regulator of the cell cycle. Under normal conditions, pRB prevents cells from progressing from the G1 phase to the S phase by binding to and inhibiting the activity of the E2F transcription factor. This mechanism ensures that cells do not replicate their DNA until they are fully prepared for division. When both copies of the RB1 gene are mutated or inactivated, pRB function is lost. Consequently, the cell cycle proceeds unchecked, leading to unregulated DNA synthesis and uncontrolled cellular proliferation.
Retinoblastoma development follows Alfred Knudson's 2-hit hypothesis. According to this model, the 1st hit involves the inheritance of a defective RB1 allele as a germline mutation in hereditary cases. In sporadic cases, the 1st mutation occurs somatically in retinal cells. The 2nd hit involves a somatic mutation that inactivates the 2nd RB1 allele, leading to tumorigenesis. This 2-hit model explains why hereditary retinoblastoma typically presents earlier and involves bilateral or multifocal tumors, whereas sporadic cases tend to present later and are usually unilateral.
In addition to RB1 mutations, a minority of retinoblastomas develop through pathways independent of this gene. These cases are often driven by alternative genetic alterations, one of which is the amplification of the MYCN oncogene, which can drive aggressive tumor growth independent of RB1 inactivation. In other cases, RB1 function is lost through epigenetic silencing, such as hypermethylation of its promoter, leading to reduced gene expression.[34]
Loss of pRB function releases the E2F transcription factor, which activates genes necessary for DNA replication and cell division. This unchecked activation of the cell cycle machinery allows retinal progenitor cells to proliferate uncontrollably.[35]
In addition to promoting cell cycle progression, RB1 inactivation disrupts mechanisms that maintain chromosomal stability. This disruption leads to the accumulation of additional genetic mutations and chromosomal abnormalities. These secondary alterations contribute to tumor progression and heterogeneity.
Normal retinal cells undergo programmed cell death to eliminate damaged or unwanted cells. However, in retinoblastoma, mutations in the RB1 gene impair apoptotic pathways, allowing abnormal cells to survive and proliferate. Furthermore, tumor cells may upregulate antiapoptotic proteins like Bcl-2 or activate survival signaling pathways, including PI3K/Akt, enhancing their ability to evade cell death.[36]
As the tumor grows, it requires the development of new blood vessels to supply oxygen and nutrients. Retinoblastoma cells secrete angiogenic factors, such as VEGF, to promote neovascularization, enabling rapid tumor expansion and further supporting tumor survival and progression.[37]
Cellular Origin of Retinoblastoma
Retinoblastoma originates from immature retinal progenitor cells in early childhood. These cells have a high proliferative potential, making them more susceptible to the effects of RB1 loss. The tumor typically develops during retinal formation, which explains why it predominantly affects young children and is rare in adults.[38]
Tumor Growth and Spread
Retinoblastoma primarily grows within the eye, leading to several consequences. The tumor disrupts the retinal architecture, impairing normal retinal function. Tumor cells can also spread into the vitreous cavity, a process known as vitreous seeding. Alternatively, the neoplastic cells may also disseminate into the subretinal space.[39]
In more advanced cases, retinoblastoma can invade beyond the eye. Optic nerve invasion occurs when tumor cells spread along the optic nerve, potentially reaching the brain. Tumor cells can also penetrate the choroid and sclera, allowing access to the systemic circulation. In some cases, retinoblastoma extends into the orbit, causing proptosis and forming an extraocular mass.
Metastasis
Retinoblastoma can metastasize to distant sites, including the central nervous system (CNS, via the optic nerve), bone marrow, and lymph nodes. Extraocular spread is associated with a poor prognosis.[40]
Clinical Manifestations Linked to Pathophysiology
The clinical signs of retinoblastoma are directly related to its underlying pathophysiological processes. Leukocoria, or the white pupillary reflex, is caused by the tumor's light reflection. Strabismus may occur due to poor vision in the affected eye, often resulting from retinal detachment or tumor growth. Proptosis, or bulging of the eye, is seen in cases where the tumor invades the orbit. Pain and redness can result from secondary glaucoma or inflammation, which are common in the advanced stages of the disease.[41]
Table 3. Pathophysiology of Hereditary vs. Sporadic Retinoblastoma
|
Points of Comparison |
Hereditary Retinoblastoma |
Sporadic Retinoblastoma |
|
Genetic Mutation |
Germline RB1 mutation (present in all cells) |
Somatic RB1 mutations (occur only in retinal cells) |
|
Tumor Laterality |
Bilateral or multifocal |
Unilateral and unifocal |
|
Age of Onset |
Earlier (median age: 12 months) |
Later (median age: 24 months) |
|
Risk of Secondary Cancers |
High risk of secondary malignancies (eg, osteosarcoma) |
No increased risk of secondary malignancies |
Emerging Insights in Retinoblastoma Pathophysiology
Recent advances have provided new insights into retinoblastoma biology. The first is epigenetic regulation, as studies suggest that retinoblastoma cells exhibit widespread epigenetic changes, including DNA methylation and histone modifications, which may contribute to tumor progression. The second is the tumor microenvironment. The interaction between tumor cells and their microenvironment, including immune cells and stromal components, plays a critical role in disease progression and treatment resistance. The third is new information obtained from genomic studies, which have identified additional genetic alterations, such as mutations in MYCN, which may drive tumorigenesis in RB1-independent cases.
The pathophysiology of retinoblastoma revolves around genetic mutations in the RB1 gene and subsequent disruptions in cell cycle regulation, apoptosis, and genomic stability. Understanding these molecular mechanisms has been pivotal in developing diagnostic and therapeutic strategies for this pediatric malignancy. Ongoing research into alternative pathways, such as MYCN amplification and epigenetic modifications, holds promise for further advancements in retinoblastoma management.[42]
Histopathology
Histopathological examination of retinoblastoma provides critical insights into its cellular origin, tumor architecture, differentiation patterns, and progression. These features are not only important for confirming the diagnosis but also for assessing prognosis and guiding treatment.[43] Below is a detailed explanation of the histopathological features of retinoblastoma.
Gross Pathology
Retinoblastoma typically presents as a white, friable intraocular mass arising from the retina. The tumor may exhibit areas of calcification, necrosis, and hemorrhage. Depending on the disease stage, the tumor may be confined to the retina or extend to other ocular structures, such as the vitreous cavity, which is associated with vitreous seeding, and the subretinal space, which is linked to subretinal seeding and retinal detachment. In advanced cases, the tumor may invade the optic nerve and beyond, indicating extraocular or metastatic spread.
Microscopic Features
Retinoblastoma is composed of poorly differentiated small, round, blue cells that resemble immature retinal cells. These cells have hyperchromatic nuclei and scant cytoplasm, resulting in a high nuclear-to-cytoplasmic ratio. Mitoses are frequently observed, reflecting the tumor's high proliferative activity.[44]
Retinoblastomas often exhibit characteristic rosette formations, which represent the tumor cells' attempts at differentiation. The most characteristic type is the Flexner-Wintersteiner rosette, in which tumor cells are arranged in a circular fashion around a central lumen, mimicking the photoreceptor layer of the retina. These rosettes reflect photoreceptor differentiation. Homer Wright rosettes are also found, where tumor cells form a circular pattern around a central core of fibrillary material. These rosettes are seen in neuroectodermal tumors and indicate primitive neural differentiation. Fluerette patterns are characterized by loosely formed clusters of cells resembling photoreceptors, which suggest a higher degree of differentiation.[45]
Tumor necrosis is common in retinoblastoma due to rapid tumor growth and insufficient vascular supply. Necrotic areas may contain ghost cells, which are remnants of necrotic tumor cells. Calcification frequently occurs in necrotic areas and is considered a hallmark feature of retinoblastoma. These calcifications are often detectable through imaging studies, such as computed tomography (CT) scans, which assist in diagnosis.[46]
Retinoblastoma tumors also exhibit prominent angiogenesis, with thin-walled blood vessels permeating the tumor. This vascular proliferation supports the tumor's rapid growth but may also contribute to hemorrhagic tendencies.[47]
The differentiation of retinoblastoma tumors plays a significant role in prognosis. Well-differentiated tumors typically exhibit abundant Flexner-Wintersteiner rosettes and Fluerette patterns and are associated with a better prognosis. In contrast, poorly differentiated tumors lack rosettes and display sheets of undifferentiated small round cells. These tumors are characterized by higher proliferative activity and more aggressive clinical behavior.[48]
Invasion and Extension
Tumor extension into the optic nerve is a critical prognostic factor in retinoblastoma. Histopathological examination assesses for the presence of prelaminar invasion, which is confined to the nerve head, and postlaminar invasion, where the tumor extends beyond the lamina cribrosa. The presence of tumor cells at the optic nerve resection margin indicates a high risk of systemic metastasis.[49] Infiltration into the choroid also signals advanced disease and is associated with a poorer prognosis. Choroidal invasion can be either focal, meaning limited to a small area, or massive, indicating extensive involvement.[50]
Advanced tumors may invade the sclera, orbit, or anterior segment of the eye. Extraocular spread significantly increases the risk of metastasis to distant sites, such as the brain and bone marrow.[51] Additionally, tumor cells may exfoliate into the vitreous cavity or subretinal space. These seeding patterns complicate treatment and reduce the likelihood of successful eye salvage.[52]
Immunohistochemistry
Immunohistochemistry is often used to confirm the diagnosis of retinoblastoma and differentiate it from other small round blue cell tumors, such as medulloepithelioma and lymphoma. Key markers include the following:
- Synaptophysin: Retinoblastoma cells stain positively for this marker, indicating the tumor cells' neuroectodermal origin.
- Neuron-specific enolase: Retinoblastoma cells also exhibit positive staining for this marker, which supports retinal differentiation.
- Ki-67: The high proliferative index reflects the tumor's aggressiveness.
- S-100 Protein: This marker is positive in some cases and may suggest certain forms of differentiation.
- Glial fibrillary acidic protein: This molecule may be expressed in areas of glial differentiation within the tumor.
- Vimentin: Positive staining in undifferentiated tumor cells highlights their lack of specific differentiation.[53]
Overall, the pattern of marker expression is crucial for distinguishing retinoblastoma from other small round blue cell tumors and assessing the tumor's potential for aggressive behavior.
Differential Diagnosis
Histopathological evaluation is essential for differentiating retinoblastoma from other small round blue cell tumors of the eye. Medulloepithelioma, which originates from the nonpigmented ciliary epithelium, may be confused with retinoblastoma due to its similar appearance. Embryonal rhabdomyosarcoma, typically involving the orbit rather than the retina, also shares some histological features with retinoblastoma but is distinguished by its location. Primary intraocular lymphoma, which usually affects older patients and involves the uvea or vitreous rather than the retina, can also resemble retinoblastoma but is differentiated based on patient age and tumor site.[54]
Prognostic Features
Several histopathological features are associated with the prognosis of retinoblastoma. High-risk features include optic nerve invasion beyond the lamina cribrosa, massive choroidal invasion, scleral or orbital extension, and the presence of tumor cells at surgical resection margins. These factors indicate a more aggressive disease and an increased risk of metastasis. On the other hand, low-risk features include confinement to the retina or vitreous without any signs of invasion, suggesting a more favorable prognosis.
Pathological Staging
Pathological examination provides critical information for staging retinoblastoma. Intraocular disease is confined to the retina, vitreous, or subretinal space. Extraocular disease involves the optic nerve, choroid, sclera, or orbit. Metastatic disease refers to distant spread to the brain, bone marrow, or lymph nodes.[55]
Advances in Histopathology
Recent advancements have significantly enhanced the role of histopathology in retinoblastoma diagnosis. Molecular techniques, such as next-generation sequencing (NGS), allow for the detection of RB1 mutations and other genetic alterations. Liquid biopsy, involving the analysis of tumor DNA in aqueous humor or plasma, provides a noninvasive approach to diagnosis. Artificial intelligence (AI) is being used to assist in the analysis of histopathological images, improving both diagnostic accuracy and prognostication.
The histopathological evaluation of retinoblastoma provides valuable insights into tumor biology, differentiation, and the extent of invasion. Key features, such as Flexner-Wintersteiner rosettes, necrosis, and calcification, are crucial for diagnosis, while assessment for optic nerve or choroidal invasion provides important prognostic information. Advances in molecular techniques and immunohistochemistry are further enhancing our understanding of retinoblastoma, enabling more personalized treatment approaches and improved outcomes.
Toxicokinetics
Toxicokinetics examines the absorption, distribution, metabolism, and excretion of drugs or toxins, along with their adverse effects on the body. While toxicokinetics is not directly related to retinoblastoma itself, it becomes crucial when evaluating the pharmacological agents used during treatment, particularly chemotherapeutic drugs. Below is a detailed overview of the toxicokinetics of retinoblastoma therapies, including both systemic and localized chemotherapies.[56]
Key Chemotherapeutic Agents Used in Retinoblastoma
Several agents are employed in retinoblastoma treatment, either alone or in combination. The toxicokinetics of these agents guide dosing regimens, minimize toxicity, and enhance therapeutic efficacy. Common agents include the following:
- Vincristine, a vinca alkaloid
- Carboplatin, a platinum-based alkylating agent
- Etoposide, a topoisomerase II inhibitor
- Melphalan, an alkylating agent used during IAC
- Topotecan, a topoisomerase I inhibitor [57]
Absorption
Chemotherapeutic agents used in retinoblastoma treatment are administered through different routes to optimize drug delivery. For systemic administration, intravenous delivery of drugs such as vincristine, carboplatin, and etoposide ensures rapid and complete bioavailability. This method bypasses 1st-pass metabolism, allowing for immediate systemic distribution of the drugs throughout the body.
In localized administration, IAC delivers drugs directly to the ophthalmic artery, achieving high local concentrations in the affected area while minimizing systemic exposure. IVC, used for vitreous seeding, involves direct injection into the vitreous humor. This targeted delivery ensures that the drug reaches high local concentrations within the eye.[58]
Distribution
The distribution of chemotherapeutic agents in the body is influenced by their physicochemical properties, protein binding, and ability to penetrate ocular and systemic tissues. Vincristine is widely distributed across tissues but has limited penetration into the CNS due to its poor ability to cross the blood-brain barrier. Carboplatin exhibits moderate protein binding and is distributed throughout the extracellular fluid. When administered intraarterially, this agent can penetrate the vitreous humor effectively. Melphalan and topotecan achieve high intraocular concentrations when delivered via intraarterial or intravitreal routes. The hydrophilic nature of these agents ensures minimal systemic distribution, which helps reduce the risk of systemic toxicity.[59]
Metabolism
The metabolism of chemotherapeutic agents varies. Vincristine is metabolized by hepatic cytochrome P450 enzymes, particularly CYP3A4, into both active and inactive metabolites. Hepatic dysfunction can prolong its half-life and increase the risk of toxicity. Carboplatin, unlike cisplatin, undergoes minimal hepatic metabolism, primarily being hydrolyzed in plasma to form reactive intermediates that bind to DNA and proteins. Etoposide is metabolized in the liver through cytochrome P450 enzymes (CYP3A4 and CYP3A5), resulting in less active metabolite production. High doses or liver dysfunction may elevate toxicity. Melphalan undergoes spontaneous hydrolysis in plasma, forming active alkylating agents. The metabolism of this drug is not enzyme-dependent, which makes it relatively predictable.[60]
Elimination
Elimination of chemotherapeutic agents involves both renal and biliary pathways. The half-lives of these drugs vary depending on their metabolism and excretion.
Carboplatin is predominantly excreted unchanged through the kidneys. Clearance depends on the glomerular filtration rate (GFR), requiring dose adjustments in cases of renal impairment. Etoposide and topotecan also undergo renal elimination, with a significant portion excreted as unchanged drugs or metabolites. In contrast, vincristine is primarily eliminated through biliary excretion, with minimal renal clearance. Hepatic dysfunction can lead to drug accumulation, increasing the risk of neurotoxicity.
Vincristine has a half-life of 19 to 22 hours. Carboplatin has a half-life of 2 to 6 hours, depending on renal function. Etoposide's half-life is 4 to 11 hours.[61]
Toxicity and Adverse Effects
Toxicokinetics is crucial for understanding the toxic profiles of these agents and their dose-limiting effects.[62] Vincristine can cause neurotoxicity, including peripheral neuropathy and autonomic dysfunction (eg, constipation), which are dose-limiting toxicities. Impaired hepatic metabolism can lead to the accumulation of the drug, exacerbating these effects. Leukopenia is uncommon compared to other agents, but myelosuppression can still occur.[63] Carboplatin's dose-limiting toxicities are related to its myelosuppressive effects, primarily thrombocytopenia, anemia, and neutropenia. Nephrotoxicity, though rare, may be significant in patients with preexisting renal impairment. Ototoxicity is a concern, particularly in pediatric patients, requiring baseline and follow-up audiometry to monitor hearing function.[64]
Etoposide is most notable for its dose-limiting neutropenia, which is a significant adverse effect. Additionally, prolonged use of this drug increases the risk of secondary malignancies, particularly therapy-related acute myeloid leukemia (AML).[65] Melphalan can cause retinal toxicity when delivered at high local concentrations intraarterially. However, the localized administration limits the drug's systemic toxicity.[66] Topotecan may also cause myelosuppression, with dose-limiting neutropenia being a common concern. Renal toxicity requires dose adjustment in patients with renal dysfunction.[67]
Implications of Toxicokinetics in Retinoblastoma Treatment
Dosing strategies for retinoblastoma therapies are designed to optimize efficacy while minimizing toxicity. Systemic chemotherapy doses are typically calculated based on body surface area (BSA) or weight, as these measurements help tailor the treatment to the individual patient and reduce the risk of adverse effects. In addition, pharmacokinetically guided dosing is used for certain agents, such as carboplatin. This approach takes renal function, specifically the glomerular filtration rate, into account to ensure an appropriate balance between therapeutic efficacy and toxicity mitigation.[68]
Localized delivery methods are essential in enhancing drug efficacy while minimizing systemic side effects in retinoblastoma treatment. IAC achieves high intraocular drug concentrations while reducing systemic exposure, minimizing adverse effects like myelosuppression and nephrotoxicity. IVC directly delivers drugs into the vitreous cavity, bypassing systemic circulation and the blood-retinal barrier.[69]
Combination therapy, which involves using multiple agents with different toxicokinetic profiles, allows for the optimization of drug dosing. This approach reduces systemic toxicity and enhances tumor eradication by targeting the cancer through various mechanisms.
However, individual variability in toxicokinetics must also be considered. For instance, patient age is critical, as neonates and young children have immature hepatic and renal systems that can alter drug metabolism and elimination. Genetic variabilities, such as polymorphisms in cytochrome P450 enzymes (eg, CYP3A4) and drug transporters (eg, P-glycoprotein), can also affect drug metabolism and distribution. In addition, organ dysfunction, particularly hepatic or renal impairment, necessitates dose adjustments to prevent drug accumulation and associated toxicity.
Understanding the toxicokinetics of chemotherapeutic agents used in retinoblastoma is crucial for optimizing treatment outcomes and minimizing adverse effects. The pharmacokinetics of these drugs inform dosing regimens, delivery method selection, and toxicity management, particularly in pediatric patients. Advances in pharmacokinetic modeling and localized drug delivery have significantly enhanced the therapeutic index of these agents, contributing to the improved success of retinoblastoma treatment.[70]
History and Physical
Patients with retinoblastoma typically present within the 1st year of life for bilateral disease and 3 years for unilateral disease. Inquiring about a family history of ocular malignancies is crucial. The history and physical examination are key components in evaluating suspected retinoblastoma, as they guide early detection, prompt diagnosis, and appropriate management. Below is a detailed outline of the necessary assessments during history-taking and the physical examination.[71]
History-Taking
The most common presenting features of retinoblastoma include leukocoria, strabismus, eye redness or pain, visual impairment, and proptosis (see Image. Patient with Retinoblastoma). Leukocoria is a white pupillary reflex, often reported by parents as a "white spot" in the eye when light shines on it, such as in photographs with a flash. Leukocoria occurs in 60% to 80% of cases and serves as the hallmark sign of retinoblastoma.[72] Strabismus, or misalignment of the eyes, can result from vision loss in the affected eye and is present in 20% to 30% of cases.[73]
A red or painful eye may occur in advanced cases due to secondary glaucoma, inflammation, or tumor extension.[74] Older children who can verbalize visual symptoms often report decreased vision. Proptosis, or orbital swelling, indicates advanced disease with orbital invasion.
Questions about the duration of these symptoms and whether they have been progressive should be asked. The involvement of one or both eyes can help differentiate between sporadic and hereditary retinoblastoma.[75]
Approximately 45% of retinoblastoma cases are hereditary. Clinicians should inquire about a family history of retinoblastoma, childhood eye cancers, and nonocular malignancies, which may suggest a germline RB1 mutation. A family history of osteosarcoma or soft tissue sarcomas could indicate hereditary cancer syndromes.[76][77]
Information about any prenatal, perinatal, or postnatal complications should be elicited. Delayed milestones may also be relevant in advanced disease or syndromic presentations.[78] Environmental exposures, such as radiation or mutagenic agents during pregnancy, may be linked to sporadic cases. Maternal health during pregnancy should be asked, as conditions like gestational diabetes and infections could contribute to risk.[79]
Although not causative, previous ocular trauma or infections may mimic symptoms of retinoblastoma, such as leukocoria due to retinal detachment. The history of cancer in the child or any syndromic conditions linked to RB1 mutations should also be explored.
Physical Examination
The general examination involves assessing overall health and nutritional status to identify systemic symptoms, such as weight loss, fatigue, and fever, which may indicate metastatic disease. Dysmorphic features or signs of syndromes associated with retinoblastoma, such as chromosomal abnormalities, should also be evaluated.[80]
A comprehensive ocular examination is critical in identifying retinoblastoma. The key components of this examination include external and pupil assessments.
During the external examination, the eyelids and orbital region should be inspected for signs of swelling or proptosis, which may suggest orbital extension. Additionally, any signs of infection or inflammation in advanced cases must be noted.[81]
For pupil examination, leukocoria is best observed in dim light using a direct ophthalmoscope or during red reflex testing. In the red reflex test, the red reflex in both eyes should be compared. A dull or absent reflex in 1 eye is concerning for retinoblastoma or another ocular pathology.[82]
Ocular alignment should be evaluated using the Hirschberg or alternate cover test to detect strabismus, a condition commonly observed in children with unilateral visual loss. Visual function must be assessed based on the child's age. Infants should be observed for fixation and following behavior, while older children can be assessed using age-appropriate visual acuity charts, such as Lea symbols or the Snellen chart.[83]
A slit-lamp examination is essential in cases of advanced disease. This examination may reveal iris neovascularization or tumor invasion of the anterior segment.[84]
A dilated fundus examination with indirect ophthalmoscopy is essential for identifying intraocular lesions in retinoblastoma, which typically presents as a white, chalky retinal mass with or without calcifications. Associated features should be examined, including retinal detachment, vitreous or subretinal seeding, and tumor vessels supplying the lesion. Bilateral examination is necessary to rule out multifocal or bilateral disease.[85]
The neurological examination must evaluate for signs of CNS involvement or metastasis. Papilledema, indicative of raised intracranial pressure, and cranial nerve palsies, which suggest optic nerve or orbital invasion, should be closely observed.[86]
Systemic examination is crucial for detecting metastatic spread, particularly in advanced cases. Lymph nodes should be palpated for cervical or preauricular lymphadenopathy. An abdominal examination should be conducted to rule out hepatomegaly or other signs of systemic involvement. Skeletal abnormalities must also be assessed for bone pain or masses that could suggest bone marrow metastases.[87]
Signs of secondary conditions should be carefully noted. Secondary glaucoma may be indicated by corneal edema, high intraocular pressure, or an enlarged corneal diameter (buphthalmos). Inflammatory changes, such as anterior segment inflammation (eg, pseudohypopyon), may occur in advanced disease.[88]
The differential diagnosis of retinoblastoma based on history and physical examination findings includes various clinical entities. Persistent fetal vasculature (PFV) can present with leukocoria but is typically unilateral and associated with microphthalmia. Coats disease, characterized by retinal telangiectasia and exudative retinal detachment, can mimic retinoblastoma and must be distinguished from this neoplasm. Congenital cataracts are another potential cause of leukocoria, often detected in infancy. Toxocariasis, a parasitic infection, can cause retinal granuloma and leukocoria. Retinal detachment may present with leukocoria or vision loss, particularly in cases of trauma.
A thorough history and physical examination are essential for the early diagnosis of retinoblastoma. Clinicians should maintain a high index of suspicion in children presenting with leukocoria, strabismus, or other ocular abnormalities. Timely referral for further evaluation, including imaging and genetic testing, is crucial for confirming the diagnosis and initiating appropriate treatment.[89]
Evaluation
Clinical History and Physical Examination
Clinical evaluation of retinoblastoma begins with a thorough history and physical examination. Common symptoms include leukocoria, strabismus, reduced vision, and ocular redness or pain. The duration and progression of these symptoms should be assessed, along with any family history of retinoblastoma, nonocular malignancies, or related genetic conditions, such as osteosarcoma. Perinatal history should be reviewed for prenatal exposures or complications, and any previous ocular trauma or infections must be considered, as these conditions may present with similar findings.
A physical examination includes an assessment of the patient's general appearance to identify dysmorphic features or signs of syndromes associated with retinoblastoma. Ocular evaluation begins with direct ophthalmoscopy, including a red reflex test to screen for leukocoria.[90] Strabismus and proptosis should be noted, and visual function may be assessed through fixation and following in infants or visual acuity tests in older children. A slit-lamp examination and a dilated fundus evaluation with indirect ophthalmoscopy are essential for identifying retinal masses, calcifications, and associated findings, such as retinal detachment.
In some cases, examination under anesthesia is necessary to facilitate a comprehensive assessment. This approach includes measuring corneal diameter, performing tonometry, and evaluating the anterior chamber with a hand-held slit lamp. Fundoscopy, cycloplegic refraction, and detailed documentation of findings are also conducted during this evaluation.[91]
Imaging Studies
Imaging is crucial to confirm the diagnosis, evaluate intraocular and extraocular tumor spread, and plan treatment. Imaging modalities that may be used to evaluate retinoblastoma include wide-field photography, ultrasonography, MRI, CT, and OCT.
Wide-field photography uses a specialized camera to capture high-resolution images of the retina and provide a panoramic view of the fundus. This modality allows for detailed documentation of tumor appearance, monitoring of treatment response, and assessment of disease progression over time. Wide-field photography is noninvasive and provides real-time visualization, making it valuable for serial examinations (see Image. Retinoblastoma Retina Scan.)
High-frequency B-scan ultrasonography is often the 1st-line imaging modality for structural assessment and staging of the tumor. This tool should reveal an echogenic mass with high internal reflectivity, a feature commonly associated with calcification, which is a hallmark of retinoblastoma. This technique is quick, noninvasive, and particularly useful for identifying calcifications while determining tumor size and location.[92]
MRI is the preferred method for evaluating tumor extent. This tool enhances visualization of the tumor’s relationship to the optic nerve, brain, and orbit while detecting optic nerve invasion, extraocular extension, and intracranial metastasis. Retinoblastoma-associated calcifications may appear as signal voids on MRI. A key advantage of this technique is its ability to provide excellent soft-tissue contrast without exposing the patient to radiation.[93]
CT was historically used to detect ocular calcifications but has largely been replaced by ultrasound and MRI due to concerns regarding pediatric radiation exposure. However, CT is selectively used in resource-limited settings where alternative imaging modalities are unavailable. Tumor calcifications appear as hyperdense foci on CT scans.[94]
OCT provides high-resolution cross-sectional images of the retina, making it valuable for detecting small tumors and monitoring retinal changes during treatment. The use of OCT is generally limited to cooperative older children and adults.[95]
Genetic Testing
Genetic testing is essential for confirming hereditary retinoblastoma, stratifying risk, and guiding family counseling. Genetic studies require blood samples and tumor tissue from patients and their relatives.
RB1 mutation analysis is essential for detecting germline or somatic mutations in the RB1 gene. This analysis is critical for distinguishing hereditary from sporadic retinoblastoma and helping identify at-risk family members, enabling targeted surveillance for potential secondary cancers.[96]
Testing for MYCN oncogene amplification becomes necessary in cases where RB1 mutations are not identified. This amplification is associated with nonhereditary, aggressive, unilateral retinoblastoma, making it an important factor in risk stratification.
Liquid biopsy, which analyzes cell-free DNA (cfDNA) from aqueous humor or plasma, offers a noninvasive method for tumor genotyping. This approach provides real-time insights into tumor progression or treatment response.
Pathological Examination
Pathology is an essential step following enucleation or biopsy, providing definitive diagnostic and prognostic information for retinoblastoma. During gross examination, enucleated eyes are assessed for tumor size, the presence of calcifications, and any signs of extraocular extension.[97] Microscopic examination reveals characteristic features of retinoblastoma. Tumor cells are typically small, round, and blue, with hyperchromatic nuclei. Key histological findings include Flexner-Wintersteiner rosettes, which are specific to retinoblastoma and indicate photoreceptor differentiation, and Homer Wright rosettes, which suggest primitive neural differentiation. Other notable findings may include areas of necrosis and calcification.[98]
Immunohistochemistry further aids in diagnosis. The tumor cells stain positively for synaptophysin and neuron-specific enolase, which indicates a neuroectodermal origin of the tumor. Ki-67, a marker of cell proliferation, is also commonly expressed in retinoblastoma, reflecting high proliferative activity within the tumor.[99]
Laboratory Tests
Laboratory tests are critical in monitoring patients with retinoblastoma, especially during chemotherapy, and in assessing potential metastatic disease. A complete blood count is routinely performed to evaluate bone marrow function, which is especially important during chemotherapy, as treatment can impact hematopoiesis. Renal and liver function tests are necessary to monitor organ function before and during chemotherapy. These tests ensure that the organs are functioning properly and can tolerate the treatment's side effects. In cases of suspected metastatic disease, a lumbar puncture or bone marrow biopsy may be performed to assess for CNS or systemic spread.[100]
Thus, a systemic assessment is often necessary. Systemic evaluations include a physical examination, MRI of the orbit and brain, bone scan, bone marrow aspiration, and lumbar puncture to evaluate for metastatic disease.[101]
Differential Diagnosis
The clinician must distinguish retinoblastoma from other conditions that can cause leukocoria and intraocular masses. PFV typically presents as a unilateral condition with microphthalmia. This developmental anomaly can mimic retinoblastoma but differs by its distinct ocular features. Coats disease is characterized by retinal telangiectasia and exudative retinal detachment, often presenting with leukocoria. This condition can be mistaken for retinoblastoma due to the overlap in clinical presentation. However, Coats disease differs due to its associated vascular abnormalities.
Congenital cataracts cause clouding of the lens, which can result in leukocoria. Although cataracts can appear similar to retinoblastoma, the key difference lies in the opacity of the lens, which is not a feature of retinoblastoma. Toxocariasis presents with granulomatous retinal lesions and is associated with a history of exposure to pets or contaminated soil. The lesions and clinical history help distinguish toxocariasis from retinoblastoma, which typically involves solid masses with distinct histological features.
Staging and Classification
Staging and classification systems are essential for stratifying retinoblastoma and guiding treatment decisions. Two approaches are used for these objectives.
The IIRC divides intraocular retinoblastoma into groups A to E based on tumor size, location, and seeding. Group A includes small tumors less than 3 mm in size that are confined to the retina, while group E encompasses extensive tumors that require enucleation due to their size, extraocular extension, or both.[102]
The TNM staging system is another important tool in retinoblastoma management, accounting for 3 key factors. "T" refers to tumor size and invasion, distinguishing between intraocular and extraocular involvement. "N" evaluates regional lymph node involvement, indicating potential metastatic spread to nearby lymph nodes. "M" assesses the presence of distant metastases, helping to determine the extent of systemic involvement.
Interprofessional Team Involvement
An interprofessional approach is essential for the comprehensive evaluation and management of retinoblastoma, ensuring that all aspects of care are addressed effectively. The ophthalmologist plays a central role in conducting detailed ocular assessments and managing localized treatments, such as laser therapy and cryotherapy, to target the eye tumor. The radiologist interprets imaging findings, including ultrasound, MRI, and CT scans, to assess the extent of the disease and identify any signs of extraocular spread or metastasis. The pediatric oncologist is responsible for overseeing systemic chemotherapy and coordinating care for patients with metastatic disease, ensuring that appropriate treatment regimens are followed and side effects are managed.
The geneticist conducts genetic testing, such as RB1 mutation analysis, and provides valuable family counseling to assess hereditary risks and guide surveillance for at-risk family members. The pathologist confirms the diagnosis through histopathology, identifying characteristic tumor cells and confirming the presence of key histological features.
A detailed evaluation of retinoblastoma involves integrating clinical findings, imaging studies, genetic testing, and pathology. Early and accurate diagnosis enables timely treatment, improves outcomes, and reduces complications. Advances in imaging techniques, molecular diagnostics, and interprofessional care have revolutionized the evaluation process, offering hope for better survival and quality of life for affected children.
Treatment / Management
Treatment Goals
The treatment of retinoblastoma requires an interprofessional approach focused on saving the child's life, preserving vision, and minimizing treatment-related complications. Management strategies are tailored based on factors such as tumor size, laterality (unilateral or bilateral), disease extent (intraocular or extraocular), and individual considerations like age and genetic predisposition.
The primary goal of treatment is to save the child's life by eradicating the tumor. The secondary goal is to preserve vision and, when possible, maintain the eye. A tertiary goal is to minimize treatment-related morbidity, including the risk of secondary malignancies and adverse effects.[103]
Localized Therapies
Localized therapies address small and medium-sized tumors confined to the eye, with the primary goal of avoiding systemic toxicity. These interventions directly target the tumor to reduce or destroy it while minimizing damage to surrounding tissues.
Laser photocoagulation is commonly used for small, posteriorly located tumors. This technique uses laser energy to coagulate the blood vessels supplying the tumor, resulting in tumor necrosis. Laser photocoagulation is primarily indicated for group A tumors or small group B tumors. The procedure is minimally invasive and causes minimal systemic toxicity.[104]
Cryotherapy involves tumor destruction through cycles of freezing and thawing, often using the triple freeze-thaw technique for tumors located in the preequatorial region without deeper invasion or vitreous seeding. This procedure is most effective for small anteriorly located tumors, such as those near the ora serrata. However, cryotherapy is unsuitable for larger or posterior tumors.[105]
Plaque brachytherapy involves surgically placing a radioactive plaque, typically iodine-125 or ruthenium-106, over the tumor. This technique is used for medium-sized or recurrent tumors that are resistant to other therapies. One major advantage of this procedure is that it delivers a high radiation dose directly to the tumor while minimizing damage to surrounding tissues.[106]
Thermotherapy (transpupillary thermotherapy, TTT) utilizes heat delivered via an infrared diode laser to destroy tumor cells. Although TTT is mainly used for focal consolidation following chemotherapy, it can also be used as an isolated treatment. This procedure is typically indicated for small tumors or as an adjuvant to chemotherapy. However, TTT is ineffective for larger or diffuse tumors.[107]
Systemic Chemotherapy
Chemotherapy, also known as chemoreduction, is the cornerstone of treatment for retinoblastoma and is frequently used in combination with local therapies. Chemotherapy reduces tumors to a size that allows for more effective localized treatments.
Intravenous carboplatin, etoposide, and vincristine are commonly used in 3 to 6 cycles, depending on the retinoblastoma grade. In certain cases, single-agent carboplatin or dual-agent therapy may also be utilized, yielding favorable results, particularly for selective patients. These therapies may be used as bridging therapy to avoid more aggressive treatment options. Intravitreal melphalan is employed in cases of vitreous seeding, though it carries a small risk of extraocular dissemination. Chemoreduction is often followed by cryotherapy or TTT to maximize tumor control further.[108](A1)
Chemotherapy is particularly beneficial for reducing tumor size before initiating localized treatments, which is especially true for cases involving large tumors, typically classified as group B or C, where size reduction is needed to allow for more targeted, eye-preserving therapies.
The most common chemotherapy regimen is the combination of vincristine, carboplatin, and etoposide (VCE), which is the standard triple-drug therapy for retinoblastoma. This regimen is indicated for bilateral or multifocal disease and is essential for tumors that require size reduction before focal treatments can be applied. One of the key advantages of chemotherapy is that it reduces the tumor burden, making eye-preserving treatments more feasible, and it also helps reduce the risk of systemic spread. However, the treatment carries several potential adverse effects, including myelosuppression, neurotoxicity (associated with vincristine), nephrotoxicity (due to carboplatin), and an increased risk of secondary malignancies (related to etoposide).[109]
Other Procedures
Other interventions for retinoblastoma include IAC, IVC, brachytherapy, and enucleation. These approaches aim to effectively treat tumors while minimizing systemic toxicity or preserving the eye in certain cases.
IAC involves the insertion of a catheter into the femoral artery, which is advanced toward the ophthalmic artery. This modality delivers high concentrations of chemotherapy drugs, such as melphalan, topotecan, and carboplatin, directly to the affected eye. IAC is typically indicated for advanced intraocular retinoblastoma (group C or D) and recurrent tumors. The main advantage of this approach is the high drug concentration it delivers to tumor cells while significantly reducing systemic toxicity. Additionally, IAC is highly effective for treating vitreous seeds. However, the procedure carries risks such as vascular complications, orbital edema, and retinal toxicity.
In IVC, chemotherapy drugs like melphalan and topotecan are injected directly into the vitreous cavity to address vitreous seeding. IVC is indicated for cases where vitreous seeds persist after systemic treatment or IAC. The main advantage of this procedure is that it provides a localized treatment specifically targeting vitreous seeding. However, one of the risks is the potential for extraocular spread, which may be minimized with the proper technique.[110]
Brachytherapy is used for anterior tumors that do not involve vitreous seeding or tumors that have shown resistance to chemotherapy. This targeted radiation therapy is effective for specific tumor locations.
Enucleation may be considered when vision preservation is no longer possible or the risk of extraocular spread is considerable (see Image. Enucleation). This surgical procedure removes the eye in cases of advanced intraocular disease. Enucleation is typically indicated in the presence of anterior chamber infiltration, neovascular glaucoma, invasion of the optic nerve, or cancerous involvement of more than half of the vitreous volume. Enucleation is also a preferred option when chemotherapy has failed or when the disease is diffuse, leading to poor visual prognosis and a high risk of recurrence. Additional indications include group E tumors or tumors with no potential for vision preservation, as well as eyes with secondary glaucoma, severe pain, or refractory disease.
Enucleation requires minimal manipulation and excises approximately 10 mm of the optic nerve.[111] Recent advancements in enucleation techniques allow for the removal of a long segment of the optic nerve under direct vision.[112] Enucleation prevents metastatic spread in advanced disease, but it does result in the loss of the eye and vision.(B3)
Management of Extraocular Extension
Management of extraocular extension is critical, requiring a comprehensive approach to prevent further complications. In cases of retrolaminar or massive choroidal spread following enucleation, adjuvant chemotherapy is administered for 6 months. EBRT is considered when the tumor extends to the cut end of the optic nerve at enucleation or through the sclera.[113]
Radiation therapy is an important component in managing extraocular extension. EBRT serves as a salvage treatment for recurrent or resistant tumors, as well as for extraocular spread in selected cases. However, EBRT is generally avoided when possible, particularly in hereditary retinoblastoma, due to the high risk of inducing secondary malignancies. Though the tumor cells are radiosensitive, radiation therapy can cause cataracts, radiation neuropathy, radiation retinopathy, and hypoplasia of the orbit.[114] EBRT also presents risks related to facial bone growth abnormalities and cataracts. EBRT delivers radiation to the entire orbit when necessary.
Proton beam therapy, a more precise form of radiation, is used as a more precise alternative to EBRT. This procedure offers reduced side effects, making it suitable for cases with similar radiation needs.
For advanced or metastatic disease, high-dose chemotherapy with stem cell rescue may be employed. This treatment is used in cases of extraocular or metastatic retinoblastoma. Autologous stem cell transplantation may be performed to restore bone marrow function following high-dose chemotherapy. In situations where the disease is widespread, palliative care becomes the focus, shifting from curative treatments to symptom management and improvement of the patient’s quality of life.[115]
Interprofessional Approach
Effective management of retinoblastoma requires a collaborative effort from a range of specialists to ensure comprehensive care. Ophthalmologists play a crucial role in diagnosing the disease and managing ocular treatments. Oncologists administer systemic chemotherapy and closely monitor for systemic complications that may arise during treatment.
Radiologists contribute by performing essential imaging studies to stage the disease and track treatment progress, helping guide the management plan. Geneticists are key in providing genetic counseling and risk assessment, especially for patients with hereditary retinoblastoma. Pathologists are responsible for confirming the diagnosis and evaluating prognostic factors, particularly in enucleated eyes, to guide treatment decisions.
Table 4. Treatment by Staging
|
Tumor Classification |
Treatments |
|
Groups A and B |
Focal therapies (laser, cryotherapy, TTT) ± systemic chemotherapy |
|
Group C |
Systemic chemotherapy + focal therapies |
|
Group D |
Systemic chemotherapy, IAC, ± IVC |
|
Group E |
Enucleation ± systemic chemotherapy for high-risk features |
|
Extraocular |
Systemic chemotherapy, radiation therapy, ± high-dose chemotherapy with stem cell rescue |
|
Metastatic |
High-dose chemotherapy, stem cell rescue, or palliative care |
Surveillance and Follow-Up
In addition to treatment, careful follow-up at repeated intervals is necessary to detect any recurrence or the development of new tumors early, particularly in patients with inherited disease. This ongoing monitoring helps ensure the best possible outcome for the patient.
Frequent eye examinations, often under anesthesia for younger children, are performed to monitor for recurrence or new tumor formation. Genetic counseling and testing are essential for hereditary cases, with RB1 mutations being tested and family members, including siblings, undergoing screening when appropriate. Long-term monitoring focuses on surveillance for secondary malignancies, especially in hereditary cases. Surveillance should include annual physical examinations, imaging, and ophthalmologic follow-ups to provide comprehensive care and detect potential complications early.[116]
Advances in Treatment
Advances in retinoblastoma treatment include investigational approaches like gene therapy, which targets the RB1 pathway or associated signaling pathways. Targeted therapies, such as VEGF inhibitors, aim to limit angiogenesis and inhibit tumor growth. Liquid biopsy is another emerging tool, allowing for the monitoring of tumor-derived DNA in aqueous humor and providing a real-time assessment of disease status.[117]
The management of retinoblastoma is tailored to the disease stage and patient-specific factors, with a focus on preserving life, salvaging vision, and minimizing adverse effects. Advances in chemotherapy delivery, genetic understanding, and focal therapies have contributed to improved outcomes. However, challenges remain in addressing care disparities between high- and low-resource settings. Continued innovation and collaboration are essential to achieving better outcomes for all children with retinoblastoma.[118]
Differential Diagnosis
Retinoblastoma is the most common intraocular malignancy in children, but several other ocular and systemic conditions can mimic its manifestations, particularly leukocoria. The following is a detailed list of differential diagnoses for retinoblastoma, organized by shared clinical features and distinguishing characteristics.
Conditions Causing Leukocoria
Leukocoria is a hallmark sign of retinoblastoma. However, several other conditions can also present with a white pupillary reflex.
PFV is a congenital anomaly resulting from incomplete regression of the fetal hyaloid vascular system. This condition is typically unilateral and associated with microphthalmia (smaller eye size), a shallow anterior chamber, and elongated ciliary processes. Unlike retinoblastoma, PFV lacks calcifications and retinal masses on imaging. Ultrasonography reveals a stalk of tissue extending from the optic nerve to the posterior lens capsule.[119]
Coats disease involves idiopathic retinal telangiectasia and exudative retinal detachment. This pathology presents unilaterally in 90% of cases. Retinal telangiectasia and yellow subretinal exudates are often evident on examination. Coats disease may mimic retinoblastoma, as it manifests with exudative detachment and leukocoria. However, unlike retinoblastoma, imaging shows no calcifications, and fundoscopy reveals dilated and tortuous retinal vessels.[120]
Congenital cataracts cause opacification of the lens, resulting in a white pupillary reflex. These cataracts may be bilateral or unilateral, but they do not involve the retina or form a mass. Distinguishing features include a clear absence of retinal masses and calcifications on imaging, with leukocoria being confined to the lens.
Toxocariasis is an ocular parasitic infection caused by Toxocara canis or Toxocara cati. This condition presents with retinal granulomas, which appear as white lesions and may be associated with vitreous inflammation, strabismus, or decreased vision. Distinguishing features include an elevated eosinophil count or positive serology result for Toxocara, with granulomas visible on imaging but without calcifications.[121]
Retinopathy of prematurity (ROP) results from abnormal retinal vascular development in premature infants. ROP is typically bilateral, causing abnormal retinal vasculature, which can lead to retinal detachment and leukocoria in advanced stages. A history of prematurity and oxygen supplementation distinguish this condition from retinoblastoma. Imaging may show retinal folds and avascular zones but no calcifications.[122]
Familial exudative vitreoretinopathy is a genetic disorder that causes abnormal retinal vascular development. This condition often manifests with bilateral retinal detachment and leukocoria. Strabismus or vision loss may also occur. Distinguishing features include a positive family history and the absence of calcifications and tumor masses on imaging.[123]
Intraocular Tumors
Intraocular tumors may sometimes mimic the mass effect seen in retinoblastoma. One notable example is medulloepithelioma, a rare embryonal tumor that typically arises from the ciliary body. This neoplasm often presents unilaterally and can cause leukocoria, secondary glaucoma, or abnormalities in the anterior chamber. Unlike retinoblastoma, medulloepithelioma tumors originate from the ciliary body, not the retina, and do not exhibit retinal calcifications on imaging.[124]
Retinal astrocytic hamartomas are benign glial tumors. These intraocular lesions may also be confused with retinoblastoma. Retinal astrocytic hamartomas are most commonly associated with tuberous sclerosis and neurofibromatosis, presenting as yellow-white retinal masses. These neoplasms are often linked with other systemic features, such as seizures and cutaneous findings. Calcifications are sometimes observed but tend to remain stable over time. These tumors do not exhibit progressive growth, differentiating them from retinoblastoma.[125]
Systemic and Congenital Conditions
Certain systemic diseases or congenital abnormalities may present with ocular findings resembling those of retinoblastoma, making careful assessment essential for accurate diagnosis. Norrie disease is a rare X-linked recessive disorder that causes retinal dysplasia, typically leading to bilateral blindness from birth. The condition is characterized by progressive retinal detachment and leukocoria. Norrie disease is distinguished by its association with hearing loss and intellectual disability, features not seen in retinoblastoma.[126]
Coloboma is a congenital defect in the ocular structure that can cause leukocoria if it affects the macula or retina. This condition may be associated with systemic syndromes. Unlike retinoblastoma, coloboma does not present with calcifications or a retinal mass on imaging. A keyhole-shaped defect in the iris or retina is a distinguishing feature.[127]
Vitreous hemorrhage, often secondary to trauma, ROP, or Coats disease, may also present with leukocoria due to blood obscuring the fundus. This condition can reduce vision or cause strabismus but lacks evidence of a mass or calcifications on imaging. Ultrasonography reveals a mobile, echogenic vitreous, which helps differentiate vitreous hemorrhage from retinoblastoma.[128]
Infectious and Inflammatory Conditions
Some infections and inflammatory conditions can mimic retinoblastoma by causing retinal or vitreous lesions. Endophthalmitis is an intraocular infection that involves the vitreous and retina, often presenting with a red, painful eye and vision loss. Clinical signs such as hypopyon or vitreous opacities may be present. Endophthalmitis is typically distinguished by a history of surgery, trauma, or systemic infection and lacks the calcifications or mass effects seen in retinoblastoma.[129]
Uveitis is a uveal tract inflammation that can cause retinal lesions, which may produce leukocoria, retinal detachment, or granulomas. This condition is often associated with systemic autoimmune diseases. Unlike retinoblastoma, uveitis is characterized by elevated inflammatory markers, such as antinuclear antibodies (ANA) and human leukocyte antigen B27 (HLA-B27), and lacks retinal calcifications.[130]
Approach to Differentiating Retinoblastoma from Similar Clinical Entities
The diagnostic approach to differentiating retinoblastoma involves a combination of clinical examination, imaging studies, genetic testing, and laboratory tests. A thorough fundoscopic examination is essential to identify abnormalities, such as retinal masses and calcifications. Imaging studies, such as ultrasound, are useful for detecting calcifications and a well-defined mass typically seen in retinoblastoma. MRI helps distinguish retinoblastoma from other conditions causing leukocoria, such as Coats disease and PFV. Although CT is rarely used, it can detect calcifications specific to retinoblastoma.
Genetic testing plays a key role in confirming hereditary retinoblastoma by identifying mutations in the RB1 gene. Laboratory tests, including specific serologies (such as Toxocara antibodies) and inflammatory markers, help rule out infections or autoimmune conditions.
The differential diagnosis of retinoblastoma is broad, encompassing benign, malignant, and inflammatory conditions. Accurate diagnosis, achieved through a combination of clinical evaluation, imaging, and sometimes genetic or laboratory studies, is crucial for initiating appropriate treatment and avoiding unnecessary interventions.
Pertinent Studies and Ongoing Trials
Extensive research and clinical trials have been conducted on retinoblastoma, with the goal of improving diagnostic techniques, treatment protocols, and patient outcomes. This section provides an overview of significant studies and ongoing clinical trials in this field.
Pertinent studies include the following:
-
Retinoblastoma Outcomes Based on the 8th Edition American Joint Committee on Cancer (AJCC) Staging System: A study published in Ophthalmology evaluated retinoblastoma outcomes using the 8th edition AJCC classification. The research highlighted the system's effectiveness in predicting global salvage and metastasis-related mortality, demonstrating its clinical utility. (AAO Journal)
-
Global Retinoblastoma Treatment Outcomes: An international study analyzed treatment outcomes across different regions, revealing disparities in survival rates and treatment efficacy. The findings underscored the importance of standardized treatment protocols and proper resource allocation to enhance global retinoblastoma care. (AAO Journal)
-
Adjuvant Chemotherapy Cycles for High-Risk Retinoblastoma: Research published in JAMA compared the efficacy of 3 versus 6 cycles of adjuvant chemotherapy (VCE) in patients with high-risk retinoblastoma postenucleation. The study aimed to identify the optimal number of chemotherapy cycles to balance VCE's effectiveness and potential toxicity.[131] (JAMA Network)
Ongoing clinical trials include the following:
-
Intravitreal Melphalan for Retinoblastoma: A Phase II trial is investigating the safety and efficacy of adding intravitreal melphalan to standard chemotherapy for early treatment of retinoblastoma with vitreous seeds. This approach seeks to improve local tumor control while minimizing systemic toxicity.[132] (Comprehensive Cancer Information)
-
Retinoblastoma Patient Clinical Database and Tissue Repository: The Mayo Clinic is conducting a study to establish a comprehensive clinical database and tissue repository for retinoblastoma patients. The goal is to systematically record clinical presentations, treatments, and outcomes, facilitating future research and personalized treatment strategies.[133] (Mayo Clinic)
-
SJRET6 Combination Chemotherapy for Intraocular Retinoblastoma: St. Jude Children's Research Hospital is conducting a Phase II clinical trial (SJRET6) for children with newly diagnosed intraocular retinoblastoma. The study evaluates the effectiveness of combination chemotherapy in treating retinoblastoma confined to the eye. (St. Jude Children's Research Hospital)
-
Targeting NUDT21 siRNA Drugs in Retinoblastoma and Refractory Retinoblastoma: A clinical trial is exploring the use of siRNA drugs targeting NUDT21 in patients with retinoblastoma, assessing the safety and therapeutic potential of this novel treatment approach.[134] (ICHGCP)
-
Study on Retinoblastoma Treatment Using Etoposide, Carboplatin, and Melphalan: This clinical trial focuses on the conservative treatment of retinoblastoma using a combination of etoposide, carboplatin, and melphalan for patients with specific eye conditions, aiming to preserve vision and control the disease.[135] (Clinical Trials EU)
Ongoing research and clinical trials are pivotal in understanding and managing retinoblastoma. These studies aim to refine treatment protocols, reduce adverse effects, and improve survival rates, offering hope for better outcomes for affected children worldwide.[136]
Treatment Planning
Treatment planning for retinoblastoma requires an interprofessional, individualized approach aimed at preserving life, salvaging the eye, and maintaining vision while minimizing treatment-related complications. The planning process takes into account factors such as the disease stage, laterality (unilateral or bilateral), the patient's age, and the resources available for treatment.
Evaluation Before Treatment Planning
A comprehensive evaluation is essential before developing a treatment plan for retinoblastoma. A thorough ocular examination is performed through dilated fundoscopy to assess the tumor’s location, size, and number. Among imaging studies, MRI can help evaluate potential extraocular extension, optic nerve involvement, or intracranial disease. Ultrasonography is used to detect calcifications. Genetic testing is conducted to differentiate between hereditary and sporadic cases, which guides family counseling and ongoing surveillance. Staging is determined using the IIRC or TNM system to guide treatment decisions.[137]
Factors Influencing Treatment Decisions
Factors influencing treatment decisions for retinoblastoma are multifaceted and depend on several patient- and disease-specific elements. Disease laterality plays a significant role, as unilateral cases often require more aggressive treatments, such as enucleation, while bilateral cases focus on preserving at least 1 eye with functional vision. Tumor size and location also guide the approach, with small tumors generally responsive to focal therapies, while larger tumors or those with seeding necessitate systemic chemotherapy or other advanced treatment modalities.
Another determining factor is the extent of the disease. Intraocular involvement allows for eye-preserving treatments, whereas extraocular spread calls for systemic or multimodal therapies. The patient's age and overall health are key considerations, as younger children require therapies with minimal systemic toxicity. Available resources likewise affect treatment options, with advanced treatments, such as IAC, not universally accessible.
Interprofessional Team Involvement
As mentioned, the interprofessional team involved in retinoblastoma management includes the ophthalmologist, pediatric oncologist, radiologist, pathologist, and geneticist. Collaborative approaches are necessary to optimize patient care and outcomes.[138]
Treatment Options by Stage
Treatment options for retinoblastoma vary based on disease stage, ranging from localized therapies for early disease to more aggressive approaches for advanced stages. Evaluation should involve a comprehensive ocular examination, imaging studies, and genetic testing to inform the most appropriate treatment plan.
In groups A and B, representing early disease with small, localized tumors, focal therapies such as laser photocoagulation, cryotherapy, and TTT are commonly used. Systemic chemotherapy, or chemoreduction, may be considered in cases with multifocal disease.
Groups C and D involve advanced disease with larger tumors and subretinal or vitreous seeding. Systemic chemotherapy, including VCE, is employed to reduce the tumors and manage seeding. IAC, using melphalan as an example, is an option for high-dose localized delivery, while IVC may be required for persistent vitreous seeding. Combination therapies with focal treatments are applied for residual disease.
In group E, characterized by very advanced disease with minimal potential for vision preservation, enucleation is the primary treatment to prevent systemic spread. High-risk patients may receive adjuvant chemotherapy to mitigate the risk of metastasis.
For extraocular or metastatic disease, including tumors with optic nerve extension beyond the lamina cribrosa, orbital involvement, or systemic metastasis, high-dose chemotherapy, with or without stem cell rescue, is employed. EBRT may be indicated for orbital extension, while palliative care is offered to patients with extensive metastatic disease.[139]
Treatment Modalities
As mentioned, treatment for retinoblastoma involves a range of modalities tailored to the tumor's size, location, and extent. Localized therapies, including laser photocoagulation, cryotherapy, plaque brachytherapy, and TTT, are often used for small tumors and residual disease following systemic therapy. Tumors that are large or have associated seeding are typically managed with systemic chemotherapy, commonly using the VCE regimen. Adjuvant chemotherapy may be required for high-risk histopathological features after enucleation.
IAC involves catheterizing the ophthalmic artery to deliver high drug concentrations directly to the tumor and reduce systemic side effects. IVC is utilized for vitreous seeding, with drugs such as melphalan or topotecan administered directly into the vitreous cavity.
Radiation therapy, including EBRT and proton beam therapy, may be considered for recurrent or resistant tumors. EBRT is used selectively due to its increased risk of secondary malignancies, particularly in hereditary cases. Proton beam therapy provides precise delivery with minimal damage to surrounding tissues.
Enucleation is reserved for eyes with no visual potential or those at high risk of extraocular spread. Histopathological analysis determines the need for adjuvant therapy.
Follow-Up and Surveillance
Follow-up and surveillance focus on the early detection of recurrence, monitoring for secondary malignancies, and assessment of treatment outcomes. Posttreatment ophthalmologic examinations, often under anesthesia, are conducted regularly, with MRI used to evaluate extraocular spread or intracranial involvement. Patients with hereditary retinoblastoma require lifelong surveillance for secondary cancers, such as osteosarcoma. Genetic counseling and testing play a crucial role in identifying at-risk family members who may need ongoing screening.
Advances in Treatment Planning
Advances in treatment planning that may be considered include molecular diagnostics, AI, and VEGF inhibitors. Molecular diagnostics include liquid biopsy techniques using cell-free DNA for noninvasive monitoring. AI-driven models are being developed to predict treatment responses and guide clinical decisions. VEGF inhibitors are being explored to limit angiogenesis and improve treatment outcomes.
Effective retinoblastoma treatment planning requires a personalized, interprofessional approach. Integrating clinical, imaging, and genetic data allows for tailored therapies that optimize outcomes, minimize toxicity, and preserve vision when possible. Advances in targeted therapies and precision medicine continue to enhance the prognosis for affected children.[140]
Toxicity and Adverse Effect Management
Retinoblastoma treatment is often multimodal. However, each intervention is associated with potential toxicities and adverse effects. Effective management of these complications is essential to optimize outcomes while minimizing treatment-related morbidity.
Chemotherapy-Related Toxicity
Systemic chemotherapy, including the VCE regimen, is used for chemoreduction and metastatic disease but is associated with significant complications. Myelosuppression results from bone marrow suppression, necessitating regular blood count monitoring, granulocyte colony-stimulating factor (G-CSF) for neutropenia, and transfusions for anemia or thrombocytopenia. Vincristine-induced neurotoxicity can cause peripheral neuropathy and constipation, managed through dose adjustments, supportive care, and physical therapy. Carboplatin-related nephrotoxicity may lead to renal dysfunction, requiring adequate hydration, electrolyte monitoring, and dose modifications for patients with renal impairment. Etoposide increases the risk of therapy-related AML, underscoring the need for long-term surveillance for secondary cancers.
IAC minimizes systemic toxicity but may result in local complications. Orbital edema or pain may occur due to vascular irritation, requiring symptomatic management with analgesics and close monitoring. Retinal toxicity from high drug concentrations necessitates dose adjustments and regular fundoscopic evaluations.
IVC, used for vitreous seeding, increases the risk of both local and systemic side effects. Endophthalmitis can arise from intravitreal injections, highlighting the importance of strict aseptic technique and prompt antibiotic treatment if infection occurs. Retinal detachment may result from injection-related trauma and requires regular retinal evaluations, with surgical repair if necessary.[141]
Radiation Therapy-Related Toxicity
EBRT is associated with several long-term toxicities, particularly in hereditary retinoblastoma. Secondary malignancies can arise due to radiation-induced genetic damage, necessitating efforts to minimize radiation exposure and lifelong surveillance for sarcomas and nonocular malignancies. Growth abnormalities may result from radiation effects on facial bones, with surgical correction reserved for severe deformities. Cataracts can develop due to radiation-induced lens damage, making lens-sparing techniques a priority during treatment. Cataract surgery may be required if vision deteriorates significantly.[142]
Plaque brachytherapy, though more localized, can also lead to complications. Radiation retinopathy, characterized by retinal ischemia and neovascularization, is managed with anti-VEGF injections (eg, bevacizumab) and laser photocoagulation to address ischemic areas. Optic neuropathy, caused by radiation-induced damage to the optic nerve, may be treated with corticosteroids to reduce inflammation and supportive measures to mitigate vision loss.[143]
Surgical Complications
Enucleation can lead to several postoperative complications, including orbital infections, which are managed with strict aseptic surgical techniques and antibiotic prophylaxis. Orbital implant complications, such as migration or extrusion, may require surgical revision or replacement. Beyond physical complications, the psychosocial impact of eye loss can be significant. Fitting a prosthetic eye helps restore cosmetic appearance, while psychological support and counseling play a crucial role in addressing emotional and social concerns.[144]
Long-Term Adverse Effects of Retinoblastoma and Its Management
Long-term adverse effects of retinoblastoma include an elevated risk of secondary malignancies, particularly in hereditary cases associated with RB1 mutations. Osteosarcoma, soft tissue sarcomas, and melanoma are among the most common secondary malignancies, necessitating lifelong surveillance and genetic counseling for affected families.[145] Vision loss can result from tumor invasion or treatment-related retinal and optic nerve damage, with management strategies focused on vision rehabilitation, low-vision aids, and occupational therapy.[146] The psychosocial aspects of retinoblastoma and its treatment can impact emotional well-being, body image, and quality of life, highlighting the importance of psychological counseling and support groups.[147]
Monitoring and Prevention
Regular follow-up is essential. Frequent ophthalmologic examinations help detect recurrence or complications. Regular blood work helps monitor chemotherapy-related systemic toxicity. Preventive measures include genetic testing to identify at-risk family members and patient education on recognizing early signs of complications, ensuring timely intervention and optimal long-term outcomes.
Advances in Managing Toxicities
Advances in managing toxicities focus on minimizing treatment-related morbidity while maintaining efficacy. Molecular profiling helps identify biomarkers that predict treatment response and toxicity, allowing for more personalized therapy. Targeted therapies aim to reduce systemic effects by selectively acting on cancer pathways. Noninvasive monitoring, such as liquid biopsy techniques, enables real-time assessment of treatment efficacy and toxicity, improving patient outcomes with less invasive interventions.
Effective management of the adverse effects of retinoblastoma treatment necessitates a proactive, interprofessional approach. Regular monitoring, early intervention, and supportive care are crucial for minimizing complications and enhancing the quality of life for affected children. Ongoing advancements in targeted therapies and precision medicine continue to improve both the safety and efficacy of retinoblastoma treatment.[148]
Staging
Staging is essential for guiding treatment, predicting prognosis, and standardizing care across institutions. Several staging systems are used globally, each focusing on different aspects of the disease, including intraocular and extraocular spread.
International Intraocular Retinoblastoma Classification
The IIRC is commonly used to classify intraocular retinoblastoma based on tumor size, location, and the presence of seeding. The system divides tumors into 5 groups, each indicating a different level of severity and prognosis.
Group A includes small tumors measuring 3 mm or less in size. These tumors are located away from the foveola and optic disc, and vitreous or subretinal seeding is absent. In group B, tumors exceed 3 mm in size and may be near the foveola or optic disc. However, like group A, this stage lacks vitreous or subretinal seeding. Group C describes tumors that exhibit localized seeding, either vitreous or subretinal, with the seeding confined to the vicinity of the tumor.
Group D refers to tumors with diffuse seeding, meaning large tumors with extensive vitreous or subretinal seeding. These tumors often pose greater treatment challenges. Group E encompasses advanced disease, where tumors involve more than 50% of the globe, and no visual potential remains. This group includes tumors with associated complications, such as secondary glaucoma or corneal invasion.
Table 5. International Intraocular Retinoblastoma Classification
| Group | Extent of Disease | Description |
| Group A | Small tumors | Tumors ≤ 3 mm in size; located away from the foveola and optic disc; no vitreous or subretinal seeding. |
| Group B | Larger tumors | Tumors > 3 mm in size; possibly near the foveola or optic disc; no vitreous or subretinal seeding. |
| Group C | Localized seeding | Tumors with focal vitreous or subretinal seeding; seeding confined to the tumor vicinity. |
| Group D | Diffuse seeding | Large tumors with diffuse vitreous or subretinal seeding, indicating more extensive disease |
| Group E | Advanced disease | Tumors involving > 50% of the globe; no visual potential; associated with complications like secondary glaucoma or corneal invasion |
Tumor, Node, Metastasis Staging System
The TNM staging system, developed by the AJCC, provides a comprehensive framework for staging retinoblastoma. This system incorporates both intraocular and extraocular disease features, allowing for a detailed assessment of tumor extent, regional lymph node involvement, and distant metastasis.
The tumor component (T) is classified into 4 stages. T1 refers to tumors confined to the retina, while T2 includes tumors with associated complications, such as retinal detachment or involvement of the anterior chamber. T3 is defined by extraocular extension, such as optic nerve or orbital invasion. T4 indicates the presence of distant metastasis.
In the node component (N), N0 indicates no regional lymph node involvement, whereas N1 denotes the presence of regional lymph node metastasis. The metastasis component (M) is classified as M0 for no distant metastasis and M1 for the presence of distant metastasis, including spread to the bone marrow or CNS.
Table 6. Tumor, Node, Metastasis Staging System
| Notation | Class | Description |
| T - Tumor | T1 | Tumor confined to the retina |
| T2 | Tumor with associated complications (eg, retinal detachment or anterior chamber involvement) | |
| T3 | Extraocular extension (eg, optic nerve or orbital invasion) | |
| T4 | Distant metastasis | |
| N - Node | N0 | No regional lymph node involvement |
| N1 | Regional lymph node metastasis | |
| M - Metastasis | M0 | No distant metastasis |
| M1 | Distant metastasis (eg, to the bone marrow or CNS) |
Reese-Ellsworth Classification
One of the most commonly used systems is the Reese-Ellsworth Classification, which assesses the likelihood of eye preservation following EBRT. However, the IIRC has largely replaced this system.
Group I has a very favorable prognosis, with either a solitary tumor smaller than 4 disc diameters (DD) located at or behind the equator or multiple tumors smaller than 4 DD behind the equator. Group II, associated with a favorable prognosis, includes solitary tumors between 4 and 10 DD at or behind the equator or multiple tumors of similar size behind the equator.
Group III represents a doubtful prognosis, with any lesion anterior to the equator or a solitary tumor greater than 10 DD behind the equator. Group IV, which has an unfavorable prognosis, includes multiple tumors greater than 10 DD or any lesion extending anteriorly beyond the equator. Group V is associated with a very unfavorable prognosis, encompassing massive tumors involving more than 50% of the retina or tumors with vitreous seeding.
Table 7. Reese-Ellsworth Classification
| Group | Prognosis | Description |
| Group I | Very favorable | Solitary tumor < 4 DD at or behind the equator; multiple tumors < 4 DD behind the equator |
| Group II | Favorable | Solitary tumor 4–10 DD at or behind the equator; multiple tumors 4–10 DD behind the equator |
| Group III | Doubtful | Any lesion anterior to the equator; solitary tumor > 10 DD behind the equator |
| Group IV | Unfavorable | Multiple tumors > 10 DD; any lesion extending anteriorly beyond the equator |
| Group V | Very unfavorable | Massive tumors involving > 50% of the retina; vitreous seeding |
Staging for Extraocular and Metastatic Disease
For orbital staging, optic nerve involvement and orbital extension should be assessed using imaging techniques, such as MRI or CT. This part of staging is important for planning adjuvant therapy following enucleation. For systemic staging, bone marrow biopsy and lumbar puncture are utilized to assess for distant metastases. Imaging methods, such as CT and positron emission tomography-CT (PET-CT), are employed to evaluate systemic involvement.
Staging systems, including the IIRC and TNM systems, classify retinoblastoma based on disease extent. These systems guide treatment and prognosis. Regular updates to these staging frameworks and their global adoption contribute to the standardization of care, resulting in improved outcomes for affected children.
Prognosis
Access to modern healthcare facilities is generally associated with an excellent prognosis. In developed countries, the overall survival rate exceeds 95%. The most critical risk factor associated with poor prognosis is extraocular extension, either through scleral or optic nerve invasion.
Survivors of bilateral retinoblastoma are at an increased risk of developing nonocular malignancies later in life, with the latent period for the development of a 2nd tumor typically being 9 months. EBRT reduces this latent period but also increases the risk of a 2nd malignancy within the first 30 years of life. Sarcomas are the most prevalent type of 2nd malignancy, and survival for patients who develop sarcoma is less than 50%.
The prognosis of retinoblastoma depends on multiple factors, including the stage at diagnosis, extent of the disease (ie, whether intraocular or extraocular), availability of specialized care, and genetic factors. Notably, advances in diagnostic techniques and treatment modalities have significantly improved survival rates in high-resource settings.
The disease stage plays a crucial role in determining prognosis. Intraocular retinoblastoma generally has an excellent prognosis, with survival rates exceeding 95% in high-resource countries. Eye and vision preservation depends on factors such as the tumor's location and size and the effectiveness of treatment. In contrast, extraocular retinoblastoma has a significantly worse prognosis, particularly with orbital or systemic spread. Survival rates for cases with local extraocular extension drop to 50% to 70%, while those with metastatic disease have survival rates of less than 30%.
Laterality also influences prognosis. Unilateral retinoblastoma typically has a better prognosis compared to bilateral cases, as treatment focuses on a single eye. Additionally, bilateral retinoblastoma requires more complex management due to the presence of multiple tumors and the higher risk of recurrence or new tumor development.[149]
Genetic factors are also important in prognostication. Hereditary retinoblastoma, which is associated with RB1 gene germline mutations, increases the lifetime risk of secondary malignancies, particularly sarcomas. In contrast, nonhereditary retinoblastoma does not increase the risk of secondary malignancies, and the disease's stage primarily determines prognosis.
High-risk features identified through histopathological analysis postenucleation include optic nerve invasion beyond the lamina cribrosa, massive choroidal invasion, and scleral or orbital extension. The presence of these features significantly increases the risk of metastasis, which may necessitate the use of adjuvant chemotherapy.
In HICs, the overall survival rate for retinoblastoma exceeds 95%. Prompt diagnosis and access to advanced therapies are key factors contributing to these high survival rates. In contrast, survival rates in LMICs can be as low as 50% to 60%. Delayed diagnosis, lack of resources, and limited access to specialized care are major contributors to these poorer outcomes.[150]
Vision preservation in retinoblastoma depends on various factors, including tumor location and response to treatment. Tumors located in the macula tend to have a poorer prognosis for vision. However, patients with early-stage disease (ie, groups A and B) have a higher likelihood of retaining functional vision, particularly if they respond well to focal therapies.[151]
Patients with hereditary retinoblastoma are at an increased lifetime risk of developing secondary cancers, including osteosarcoma, soft tissue sarcomas, and melanoma. This risk is further heightened by exposure to radiation therapy. Lifelong surveillance for secondary malignancies is essential to manage this increased risk.[152]
Cosmetic concerns can arise following enucleation, particularly in children, which may lead to psychosocial challenges. Fitting a prosthetic eye can help improve cosmetic outcomes, but families may also require emotional and psychological support to cope with the diagnosis and its long-term implications.[153]
Management advancements that have improved prognosis include the use of VEGF inhibitors, genetic counseling, and the early detection of recurrence or metastasis with molecular diagnostics. The prognosis of retinoblastoma is excellent for patients diagnosed early with intraocular disease and treated in high-resource settings. However, challenges remain in managing advanced or metastatic disease, particularly in low-resource settings. Continued advancements in diagnostics, treatment modalities, and global healthcare access are critical to improving outcomes for all patients with retinoblastoma.[154]
Complications
Retinoblastoma can produce the following complications if left untreated:
- Retinal detachment
- Retinal necrosis
- Orbital invasion
- Optic nerve invasion
- Blindness
- Intracranial extension
- Secondary neoplasms
- Metastasis
- Tumor recurrence
- Temporal bone hypoplasia
- Cataract
- Radiation neuropathy
- Radiation retinopathy
Complications of retinoblastoma arise from the disease itself or the adverse effects of treatments. These complications can affect vision, overall health, and long-term quality of life. Understanding and managing these complications are critical for optimizing outcomes and ensuring comprehensive care.
Disease-Related Complications
Disease-related complications may be localized or widespread. Frequent local complications include vision loss, glaucoma, and retinal detachment. Vision loss is commonly caused by macular involvement, retinal detachment, or tumor growth. The extent of vision loss depends on factors such as tumor size, location, and the patient’s treatment response. Glaucoma may result from tumor infiltration or iatrogenic injury from treatments like enucleation or radiation therapy. Symptoms of glaucoma include pain, increased intraocular pressure, and optic nerve damage. Retinal detachment, often caused by exudation from the tumor, can lead to progressive vision loss if left untreated.
Orbital extension occurs when the tumor invades the orbit, leading to proptosis and an increased risk of systemic metastasis. This manifestation is a sign of advanced disease, necessitating more aggressive treatment.[155]
Metastasis is a significant concern in retinoblastoma and is commonly observed in sites such as the bone marrow, CNS, and distant organs, including the liver. Metastasis is associated with a poor prognosis and high mortality rates.[156]
Treatment-Related Complications
Treatment-related complications include the adverse effects of chemotherapy, radiation therapy, and surgical interventions. Chemotherapy-related complications can be significant, particularly myelosuppression, which increases the risk of infections, anemia, and bleeding. These complications are typically managed with supportive care, such as granulocyte colony-stimulating factor and blood transfusions. Neurotoxicity, a common side effect of vincristine, often manifests as peripheral neuropathy and motor deficits, further complicating treatment. Etoposide increases the risk of secondary malignancies, particularly AML. Nephrotoxicity, primarily associated with carboplatin, can lead to renal dysfunction.[157]
Radiation therapy also introduces a variety of potential complications. Growth abnormalities may occur when radiation is applied to facial bones, impairing growth and causing facial asymmetry. Patients with hereditary retinoblastoma are at a heightened lifetime risk for developing secondary malignancies due to radiation exposure. Furthermore, radiation can lead to radiation retinopathy, a condition characterized by chronic ischemia and neovascularization, which can result in vision loss. Optic neuropathy, another complication, involves permanent vision impairment due to optic nerve damage.
Surgical treatments, particularly enucleation, also carry risks. Postoperative infections are common and may require antibiotic therapy to manage orbital infections. Implant extrusion, the displacement or rejection of the orbital implant, can occur and require further intervention. Additionally, the psychosocial impact of enucleation is considerable, as it may lead to cosmetic concerns and emotional distress, especially in children.[158]
Long-Term Complications
Long-term complications in patients with retinoblastoma can include secondary malignancies, psychosocial impacts, and permanent vision impairment. Hereditary retinoblastoma, in particular, increases the risk of developing secondary malignancies such as soft tissue sarcomas, osteosarcomas, and melanomas. This elevated risk persists throughout a patient's lifetime, especially for those who have undergone prior radiation therapy.
The psychosocial impact of retinoblastoma can be profound, particularly in children. Vision loss and enucleation often cause emotional and psychological challenges. Social and educational difficulties can impact patients' overall development and quality of life.[159]
Vision-related complications can be long-lasting, with many patients experiencing permanent visual impairment or blindness in 1 or both eyes. These individuals may require low-vision aids and rehabilitation services to help them adapt to their condition and enhance their independence.[160]
Management of Complications
The management of complications in retinoblastoma requires a comprehensive and interprofessional approach. Regular monitoring is essential to detect recurrence, metastasis, and secondary malignancies early, allowing for prompt intervention. Supportive care mitigates the complications of adverse effects.
Counseling helps patients and families cope with the psychological challenges associated with vision loss, enucleation, and the disease's long-term implications. Vision rehabilitation, including the use of low-vision aids and occupational therapy, is essential to enhancing the quality of life and independence of affected individuals.
The complications associated with retinoblastoma and its treatment emphasize the importance of ongoing, interprofessional care and long-term follow-up. Advances in treatment modalities continue to reduce the burden of complications, ultimately improving patient outcomes.[161]
Postoperative and Rehabilitation Care
Postoperative and rehabilitation care are essential for managing retinoblastoma, particularly for patients undergoing aggressive treatments like enucleation. This critical aspect of management focuses on physical recovery, psychological support, and optimizing quality of life.
Postoperative care for patients with retinoblastoma aims to manage immediate complications and ensure long-term monitoring. During the immediate postoperative period, close attention is given to signs of infection, bleeding, or implant displacement, with pain and inflammation managed through appropriate medications. Wound care includes maintaining proper hygiene at the surgical site and administering antibiotic ointments or eye drops to prevent infection. The stability of the orbital implant is assessed to ensure it remains properly positioned and does not extrude or migrate.
Long-term follow-up is critical to monitor for recurrence, particularly in hereditary cases where the contralateral eye may develop new tumors. Regular imaging (eg, with MRI) helps detect any residual or recurrent disease. Lifelong surveillance for secondary malignancies is essential for patients with hereditary retinoblastoma due to the increased risk of developing secondary cancers. Cosmetic outcomes are also addressed, with early prosthetic eye fitting, typically within 4 to 6 weeks postenucleation, contributing to both cosmetic and psychological well-being.
Rehabilitation care for patients with retinoblastoma is crucial for recovery and independence, addressing both physical and psychological needs. Low-vision aids and strategies to improve depth perception are recommended for patients with unilateral vision loss. Braille training, mobility aids, and orientation techniques are essential for individuals with bilateral vision loss. Referrals to specialized vision rehabilitation programs help ensure comprehensive support. Additionally, educational support is critical for patients with vision loss. These children require assistance in adapting to school environments, and advocates should push for access to necessary educational resources and technologies to facilitate learning.[162]
Psychological support is also an integral part of rehabilitation. Patients and their families often experience anxiety and depression related to vision loss or cosmetic concerns following treatment. Counseling services can help families cope with the emotional challenges that arise from the diagnosis and treatment of retinoblastoma. Peer support groups can further provide a network for families to connect with others facing similar challenges, offering mutual encouragement and shared experiences.[163]
In terms of social and developmental support, ensuring that children affected by retinoblastoma meet typical developmental milestones despite any vision impairment is crucial. Collaboration with pediatric specialists helps monitor growth and development and address any concerns. For families with hereditary retinoblastoma, genetic counseling is essential to discuss recurrence risks and establish appropriate screening protocols for siblings.
Ocular prosthesis fitting is a key aspect of postoperative care for patients with retinoblastoma who undergo enucleation. The fitting typically occurs 4 to 6 weeks after the surgery, allowing sufficient time for the surgical site to heal. In some cases, temporary conformers are initially used to maintain the shape of the orbit before the final prosthesis is fitted.
Maintaining the prosthetic eye is essential to ensure comfort and hygiene. Regular cleaning is necessary to prevent infection and discomfort. Additionally, periodic adjustments are needed, particularly in pediatric patients, to accommodate for growth and ensure the prosthesis remains functional and comfortable over time.[164]
Nutritional and physical recovery are essential. A balanced diet supports healing and strengthens the immune system, while safe physical activity promotes general health. In cases of unilateral vision loss, contact sports or activities that pose a risk to the remaining eye should be avoided to prevent further injury.[165]
Monitoring and preventive care are critical for long-term health and recovery. Genetic screening should be conducted to identify at-risk family members early, enabling timely intervention. Protective measures, such as the use of safety glasses, are necessary to prevent injury to the unaffected eye, particularly in patients with unilateral retinoblastoma. Additionally, ensuring immunization against infections is important, especially for children receiving chemotherapy.[166]
Advanced rehabilitation techniques help improve the quality of life for patients with retinoblastoma. Assistive technologies, such as screen readers and magnifiers, can help individuals with vision impairment, while occupational therapy provides training in daily living skills to promote greater independence.
Postoperative and rehabilitation care for retinoblastoma patients is crucial for achieving the best possible functional and psychological outcomes. An interprofessional approach involving ophthalmologists, oncologists, rehabilitation specialists, and mental health professionals ensures comprehensive recovery and reintegration into daily life.[167]
Consultations
The management of retinoblastoma requires a collaborative approach involving various specialists to ensure optimal outcomes. Each specialist contributes to different aspects of diagnosis, treatment, and ongoing care.
A pediatric ophthalmologist is responsible for the initial diagnosis and monitoring of tumor progression, performing detailed fundoscopic examinations under anesthesia. These specialists also administer focal therapies like laser photocoagulation and cryotherapy. Their expertise is crucial for identifying retinoblastoma and initiating primary eye-preserving treatments.[168]
Pediatric oncologists manage systemic chemotherapy and assess the potential for metastatic disease. These professionals coordinate treatment protocols, including IAC and IVC, while monitoring for systemic side effects and complications. This role is especially important for treating advanced or bilateral cases that require systemic therapy.[169]
A radiation oncologist plans and delivers radiation therapy, such as EBRT, for refractory or advanced disease. These experts minimize exposure to healthy tissues, particularly in patients with hereditary retinoblastoma. Radiation therapy is typically used when other treatments are insufficient for resistant tumors or orbital extension.[170]
A genetic counselor conducts genetic testing to identify RB1 mutations, distinguishing between hereditary and sporadic cases. These specialists provide family counseling regarding the risks of secondary malignancies and retinoblastoma occurrence in siblings or offspring. For families with hereditary retinoblastoma, the genetic counselor is essential in developing a screening plan and offering reproductive advice.[171]
A pediatric surgeon or ocular oncologist performs enucleation when eye salvage is not possible. These professionals carry out the histopathological examination of enucleated eyes to assess for high-risk features. Pediatric surgeons are essential in the surgical management of advanced retinoblastoma.[172]
Psychologists or psychiatrists address the psychological impact of retinoblastoma on children and families. These specialists provide coping strategies for dealing with enucleation, vision loss, and cosmetic concerns. Psychologists and psychiatrists offer emotional support and mental health care, contributing to the overall quality of life for patients with retinoblastoma and their families.
A rehabilitation specialist designs vision rehabilitation programs for children with partial or complete vision loss, introducing adaptive technologies and occupational therapies for daily living. These experts help children regain independence and adapt to life with visual impairment.
A radiologist performs imaging studies, including MRI and ultrasound, to evaluate tumor extent and extraocular involvement. These professionals monitor treatment response and screen for recurrence, as imaging is critical for staging and treatment planning.[173]
A pathologist analyzes enucleated specimens to identify histopathological features indicating high-risk disease and confirms the diagnosis of retinoblastoma through microscopic examination. These specialists provide prognostic information based on tumor pathology.[174]
A social worker assists families in navigating healthcare systems and accessing financial or social support, connecting them with support groups and resources. These professionals ensure families have the resources needed to cope with the emotional and financial burdens of treatment.
Consultations with an interprofessional team are essential to manage retinoblastoma effectively. Each specialist contributes unique expertise to address the diverse aspects of care, from diagnosis and treatment to rehabilitation and long-term follow-up. This collaborative approach ensures comprehensive care for patients with retinoblastoma and their families.[175]
Deterrence and Patient Education
Effective deterrence and patient education are critical components in managing retinoblastoma, particularly in preventing disease progression, improving outcomes, and ensuring long-term quality of life. Empowering families with knowledge, addressing risk factors, and emphasizing the importance of regular follow-ups are essential to these efforts.
Early Detection and Screening
Genetic counseling and testing play a vital role for families with a history of retinoblastoma. These families should undergo genetic counseling to assess the likelihood of inheritance and determine the risks to other family members. Early genetic testing of siblings and offspring helps identify individuals at risk. Regular eye examinations should be conducted for children with a family history of retinoblastoma, and newborns and infants at risk of hereditary retinoblastoma should undergo dilated fundus examinations starting at birth.[176]
Education on Disease and Symptoms
Educating parents about the early signs of retinoblastoma is vital. Symptoms such as leukocoria, strabismus, and vision loss should prompt immediate medical attention. Clinicians must provide comprehensive information about the condition, its treatment options, and expected outcomes. Early detection is key to improving survival rates and preserving vision.
Risk Reduction Strategies
Ensuring that parents understand the urgency of seeking medical attention at the first sign of symptoms is vital in preventing care delays. Additionally, families should be educated about the importance of avoiding injuries to the affected or remaining eye, particularly in children with unilateral retinoblastoma, to prevent further complications.
Adherence to Treatment and Follow-Up
Strict adherence to treatment protocols is essential for successful outcomes. Families should be encouraged to follow through with all prescribed treatments, which may include chemotherapy, radiation, or focal therapies. Regular follow-up visits are also necessary to monitor for recurrence or secondary malignancies. These visits should be scheduled more frequently in the first few years after treatment when the risk of recurrence is highest.[177]
Long-Term Surveillance
Education on the lifelong risk of secondary malignancies is crucial for families, particularly in hereditary cases of retinoblastoma. These families should be informed about the importance of regular health check-ups and imaging studies to monitor for potential secondary cancers. Families should also be instructed on the necessity of lifelong eye care, including the use of protective eyewear and avoiding high-risk activities that could lead to further injury or complications, particularly for patients who have undergone treatment for retinoblastoma.[178]
Psychosocial Support
Coping with the diagnosis of retinoblastoma can be emotionally and socially challenging for families. Psychological counseling services are essential to help them manage these difficulties. Connecting families with support groups and organizations can provide peer support and practical assistance in navigating the challenges that arise throughout the treatment process.[179]
Addressing Barriers to Care
Financial support is often a significant concern for families managing the costs of treatment. Assisting families in accessing insurance coverage or financial aid programs can help alleviate some of the financial burden. Healthcare accessibility is another critical issue, particularly in underserved regions. Advocating for improved access to specialized retinoblastoma care ensures that all patients, regardless of geographic location, have the opportunity to receive the necessary treatment and follow-up care.[180]
Parental Empowerment
Educational materials are powerful empowerment tools for parents. Easily accessible brochures, videos, and online resources that explain retinoblastoma in plain language can help families better understand the disease and its management. Equally important is teaching parents how to respond to urgent complications, such as signs of infection or vision deterioration.
Deterrence and patient education are essential for optimizing retinoblastoma outcomes. By raising awareness, promoting early detection, and ensuring adherence to treatment, healthcare providers enable families to take an active role in managing this challenging disease. A proactive approach to education and support minimizes complications, improves survival rates, and enhances the quality of life for children with retinoblastoma and their families.
Pearls and Other Issues
Retinoblastoma is a rare but highly treatable childhood malignancy. Healthcare providers should be aware of the key clinical and management points regarding this disease. These pearls and related considerations help streamline care, improve outcomes, and optimize the quality of life for affected patients and their families.
Key Pearls
- Early detection is crucial. Leukocoria is the hallmark symptom and is often the first sign noticed by parents or caregivers. Early diagnosis significantly improves survival rates and increases the likelihood of preserving vision.
- Genetic testing is important. Genetic testing for RB1 mutations should be a standard component of care for children with retinoblastoma, particularly in bilateral or familial cases. Identifying hereditary retinoblastoma enables early surveillance and preventative strategies for family members.
- Customized multimodal therapy should be provided. Treatment should be tailored to the disease stage, laterality, and individual patient factors. Treatment options range from focal therapies (eg, laser photocoagulation) for localized disease to systemic chemotherapy and enucleation for advanced stages.
- Lifelong surveillance is necessary for hereditary cases. Patients with germline RB1 mutations require ongoing monitoring for secondary malignancies, including sarcomas and melanoma. Regular follow-up visits are crucial to detect late recurrences or complications.
- Advances in conservative management help improve prognosis. Emerging therapies, such as IAC and IVC, have enhanced outcomes while reducing systemic toxicity. These methods have revolutionized treatment, enabling eye salvage in cases that previously required enucleation.
Challenges and Considerations
- Global disparities in outcomes exist. Survival rates in HICs exceed 95%, while low-resource settings report survival as low as 50%. Addressing barriers to care, such as delayed diagnosis and limited access to specialized treatment, remains a global priority.
- Retinoblastoma can have a profound psychosocial impact on patients and families. Comprehensive care should include mental health support and counseling.
- Treatment-related toxicities can be managed. Long-term complications from chemotherapy and radiation therapy require careful monitoring. Therapeutic innovations aim to reduce these risks without compromising efficacy.
- Parental education is important. Caregivers must be educated on recognizing early signs, adhering to follow-up schedules, and understanding potential complications to optimize outcomes. Awareness campaigns can improve early detection and timely referrals.
Future Directions
- Genomic and molecular advances are ongoing. Research into the molecular biology of retinoblastoma is paving the way for targeted therapies and personalized medicine. Liquid biopsy techniques may allow for less invasive disease progression and treatment response monitoring.
- AI in diagnosis shows promise. AI-powered diagnostic tools can improve early detection, particularly in resource-limited settings. Automated analysis of fundus photographs is a promising area of development.
- Improved accessibility is crucial. Expanding access to advanced therapies, such as IAC, in low-resource settings is a global healthcare priority. Training programs for healthcare providers in these regions can help bridge the gap.
Retinoblastoma management has evolved considerably over the years, with significant progress in early detection, the development of conservative treatment approaches, and the implementation of comprehensive long-term care strategies. Despite these strides, addressing global disparities in outcomes and prioritizing holistic care for patients and their families remain critical. By incorporating these key pearls into clinical practice, healthcare providers can enhance the quality and effectiveness of retinoblastoma care.
Enhancing Healthcare Team Outcomes
Retinoblastoma management is complex and requires an interprofessional team approach involving an ophthalmologist, pediatric oncologist, ocular pathologist, geneticist, allied health professionals, and parents. In most cases, the patient initially presents to a nurse practitioner or primary care provider. Since retinoblastoma is a rare condition that requires urgent attention, the patient should promptly be referred to an ophthalmologist. The cornerstone of long-term management is strict adherence to the treatment plan, along with careful follow-up at regular intervals after treatment. This follow-up is essential for the early detection of recurrence or the development of new tumors, particularly in patients with inherited disease.[181]
Patients need education about the types of treatment they will receive, including chemotherapy, surgery, and radiation. Primary care providers should be familiar with postoperative follow-up and know when to refer patients back to the ophthalmologist. As chemotherapy is the mainstay of treatment, the involvement of a pharmacist experienced in oncologic treatment is essential. The pharmacist can verify agent selection and dosing, perform medication reconciliation, and communicate any concerns back to the healthcare team. Since nurses are most likely to administer chemotherapy, maintaining a close rapport and open communication between pharmacy and nursing staff is crucial.[182]
Effective management of retinoblastoma requires close collaboration with an experienced ophthalmologist who can provide essential guidance and support. The prognosis is favorable for patients who follow their treatment plan and attend regular follow-up appointments. Given the complexity of managing this condition, an interprofessional team approach is critical. This approach involves collaboration among physicians, specialists, specialty-trained nurses, and pharmacists, all working together to achieve the best possible patient outcomes.
Media
(Click Image to Enlarge)
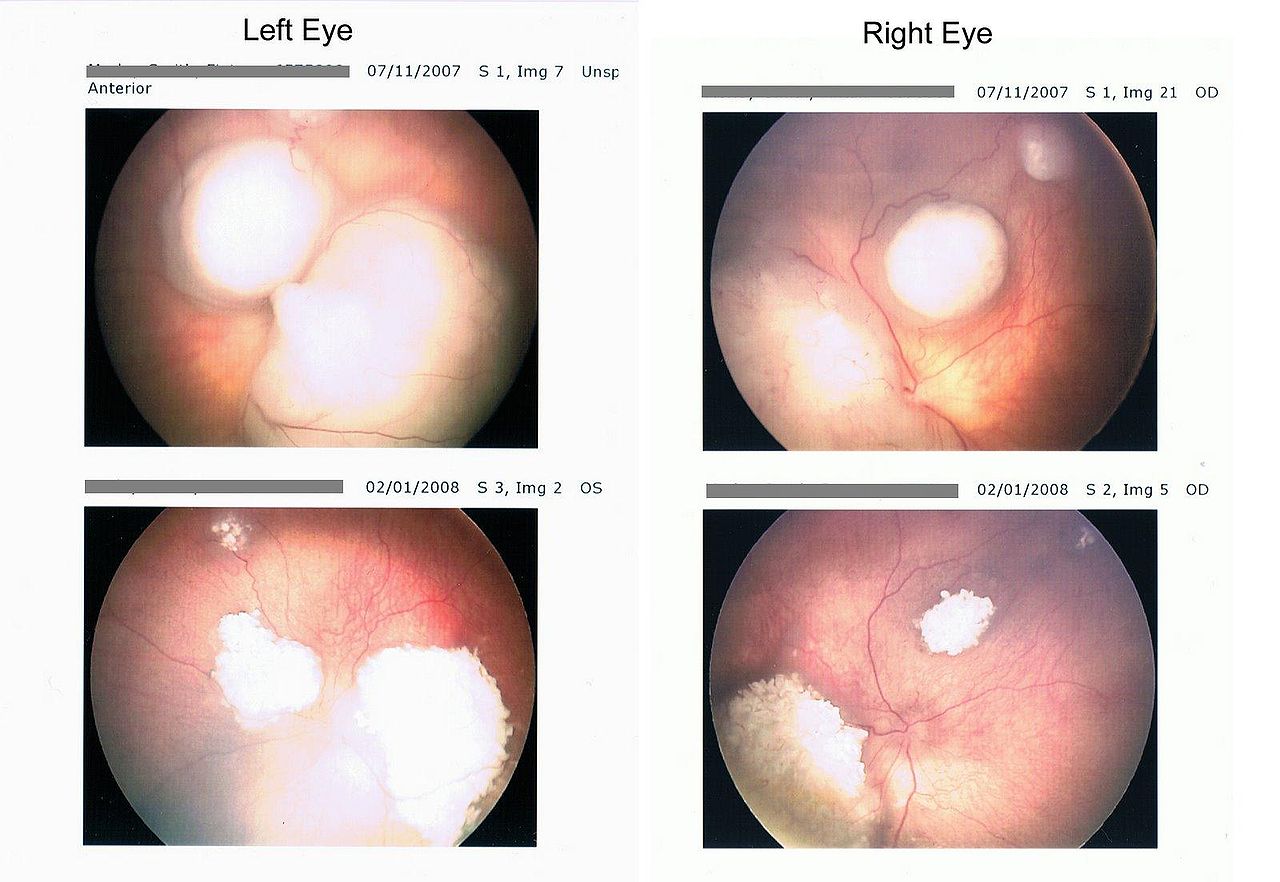
Retinoblastoma Retina Scan. This image shows bilateral, multifocal retinoblastoma tumors before and after chemotherapy. Note the size reduction in all lesions.
Contributed by Wikimedia Commons
(Click Image to Enlarge)
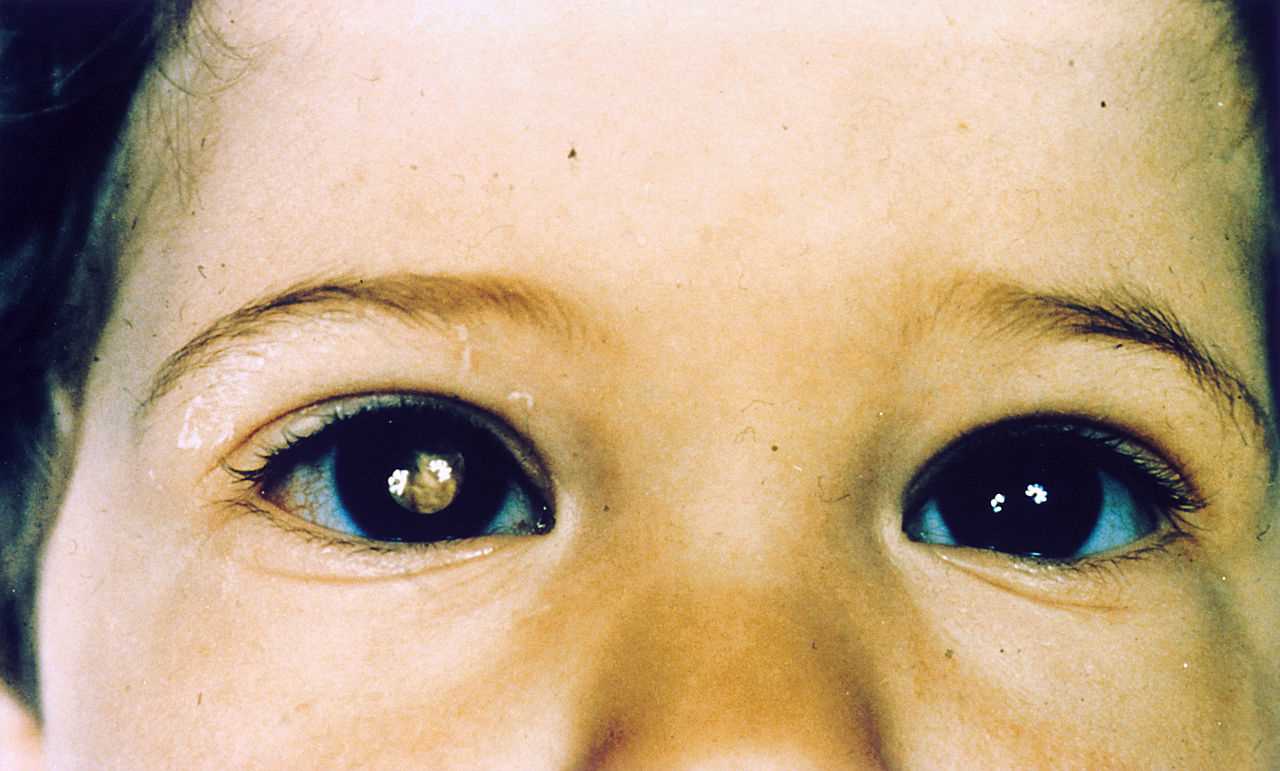
Patient with Retinoblastoma. This image shows a child with right eye leukocoria and mild injection associated with strabismus. These signs are indicative of retinoblastoma.
Contributed by Wikimedia Commons (Public Domain)
(Click Image to Enlarge)
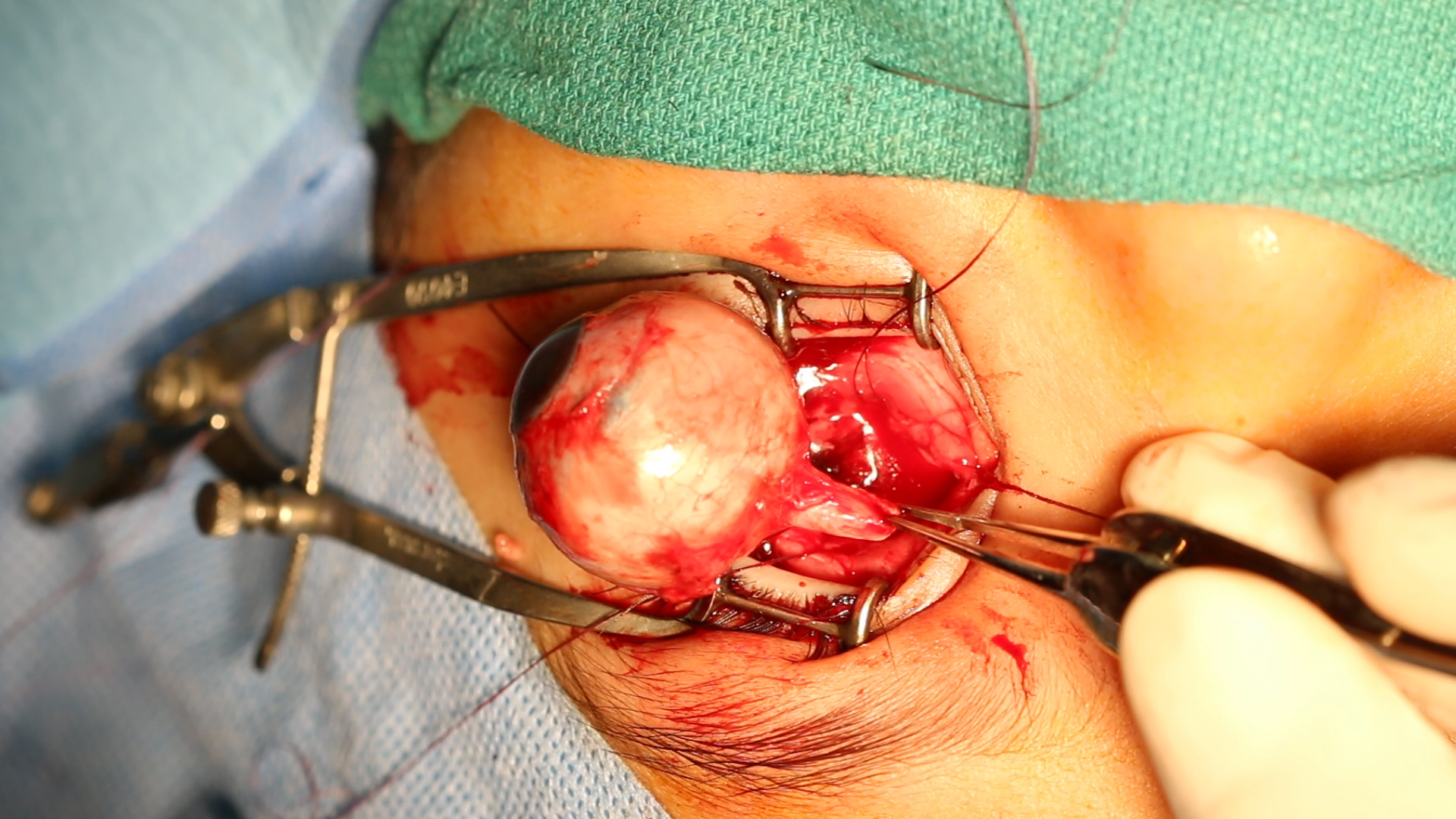
Enucleation. This image shows the enucleation of an eye using the superomedial approach to the optic nerve. This technique preserves sufficient optic nerve length, which is crucial in intraocular malignancies, particularly large retinoblastomas.
Contributed by Professor Bhupendra C. K. Patel MD, FRCS
References
Mattosinho CCS, Moura ATMS, Oigman G, Ferman SE, Grigorovski N. Time to diagnosis of retinoblastoma in Latin America: A systematic review. Pediatric hematology and oncology. 2019 Mar:36(2):55-72. doi: 10.1080/08880018.2019.1605432. Epub 2019 Apr 24 [PubMed PMID: 31014139]
Level 1 (high-level) evidenceAlkatan HM, Al Marek F, Elkhamary S. Demographics of Pediatric Orbital Lesions: A Tertiary Eye Center Experience in Saudi Arabia. Journal of epidemiology and global health. 2019 Mar:9(1):3-10. doi: 10.2991/jegh.k.181224.001. Epub [PubMed PMID: 30932383]
Kletke SN, Feng ZX, Hazrati LN, Gallie BL, Soliman SE. Clinical Predictors at Diagnosis of Low-Risk Histopathology in Unilateral Advanced Retinoblastoma. Ophthalmology. 2019 Sep:126(9):1306-1314. doi: 10.1016/j.ophtha.2019.04.003. Epub 2019 Apr 12 [PubMed PMID: 30986443]
Adam MP, Feldman J, Mirzaa GM, Pagon RA, Wallace SE, Amemiya A, Lohmann DR, Gallie BL. Retinoblastoma. GeneReviews(®). 1993:(): [PubMed PMID: 20301625]
Xu Z, Ye H, Yang H. A 57-year-old female with retinoblastoma. Eye (London, England). 2025 Jan 20:():. doi: 10.1038/s41433-025-03617-4. Epub 2025 Jan 20 [PubMed PMID: 39833578]
Cruz-Abrams O, Dodds Rojas R, Abramson DH. Machine learning demonstrates clinical utility in distinguishing retinoblastoma from pseudo retinoblastoma with RetCam images. Ophthalmic genetics. 2025 Jan 20:():1-6. doi: 10.1080/13816810.2025.2455576. Epub 2025 Jan 20 [PubMed PMID: 39834033]
Ansari M, Gupta C, Kulkarni YA, Singh K. Functionalization of polymeric nanomicelles and mixed nanomicelles for targeted retinal delivery in the management of retinoblastoma. International journal of pharmaceutics. 2025 Feb 25:671():125235. doi: 10.1016/j.ijpharm.2025.125235. Epub 2025 Jan 16 [PubMed PMID: 39826786]
Kim S, Cho CS, Jang HY, Jo DH, Kim JH. Ca(V)3.3 T-type calcium channels contribute to carboplatin resistance in retinoblastoma. The Journal of biological chemistry. 2025 Jan 16:301(2):108199. doi: 10.1016/j.jbc.2025.108199. Epub 2025 Jan 16 [PubMed PMID: 39826688]
Aschero R, Simao M, Catala-Mora J, L Chantada G. Risk Factors for Extraocular Relapse in Retinoblastoma. Seminars in ophthalmology. 2025 Jan 9:():1-11. doi: 10.1080/08820538.2025.2450682. Epub 2025 Jan 9 [PubMed PMID: 39789868]
Zahn J, Chan MP, Wang G, Patel RM, Andea AA, Bresler SC, Harms PW. Altered Rb, p16, and p53 expression is specific for porocarcinoma relative to poroma. Journal of cutaneous pathology. 2019 Sep:46(9):659-664. doi: 10.1111/cup.13480. Epub 2019 May 3 [PubMed PMID: 31012122]
Yun J, Li Y, Xu CT, Pan BR. Epidemiology and Rb1 gene of retinoblastoma. International journal of ophthalmology. 2011:4(1):103-9. doi: 10.3980/j.issn.2222-3959.2011.01.24. Epub 2011 Feb 18 [PubMed PMID: 22553621]
Li YP, Wang YT, Wang W, Zhang X, Shen RJ, Jin K, Jin LW, Jin ZB. Second hit impels oncogenesis of retinoblastoma in patient-induced pluripotent stem cell-derived retinal organoids: direct evidence for Knudson's theory. PNAS nexus. 2022 Sep:1(4):pgac162. doi: 10.1093/pnasnexus/pgac162. Epub 2022 Aug 17 [PubMed PMID: 36714839]
House RJ, Hsu ST, Thomas AS, Finn AP, Toth CA, Materin MA, Vajzovic L. Vascular Findings in a Small Retinoblastoma Tumor Using OCT Angiography. Ophthalmology. Retina. 2019 Feb:3(2):194-195. doi: 10.1016/j.oret.2018.09.018. Epub 2018 Sep 29 [PubMed PMID: 31014771]
Nag A, Khetan V. Retinoblastoma - A comprehensive review, update and recent advances. Indian journal of ophthalmology. 2024 Jun 1:72(6):778-788. doi: 10.4103/IJO.IJO_2414_23. Epub 2024 May 24 [PubMed PMID: 38804799]
Level 3 (low-level) evidenceJoseph S, Pike S, Peng CC, Brown B, Xu L, Berry JL, Chévez-Barrios P, Hubbard GB, Grossniklaus HE. Retinoblastoma with MYCN Amplification Diagnosed from Cell-Free DNA in the Aqueous Humor. Ocular oncology and pathology. 2024 Apr:10(1):15-24. doi: 10.1159/000533311. Epub 2023 Aug 25 [PubMed PMID: 38751495]
Marshall AE, Roes MV, Passos DT, DeWeerd MC, Chaikovsky AC, Sage J, Howlett CJ, Dick FA. RB1 Deletion in Retinoblastoma Protein Pathway-Disrupted Cells Results in DNA Damage and Cancer Progression. Molecular and cellular biology. 2019 Aug 15:39(16):. doi: 10.1128/MCB.00105-19. Epub 2019 Jul 29 [PubMed PMID: 31138663]
van Harn T, Foijer F, van Vugt M, Banerjee R, Yang F, Oostra A, Joenje H, te Riele H. Loss of Rb proteins causes genomic instability in the absence of mitogenic signaling. Genes & development. 2010 Jul 1:24(13):1377-88. doi: 10.1101/gad.580710. Epub 2010 Jun 15 [PubMed PMID: 20551164]
Parry MF. The in-vitro activity of azlocillin: a community hospital study of 1900 clinical isolates. The Journal of antimicrobial chemotherapy. 1983 May:11 Suppl B():15-20 [PubMed PMID: 6619025]
Mandigo AC, Shafi AA, McCann JJ, Yuan W, Laufer TS, Bogdan D, Gallagher L, Dylgjeri E, Semenova G, Vasilevskaya IA, Schiewer MJ, McNair CM, de Bono JS, Knudsen KE. Novel Oncogenic Transcription Factor Cooperation in RB-Deficient Cancer. Cancer research. 2022 Jan 15:82(2):221-234. doi: 10.1158/0008-5472.CAN-21-1159. Epub 2021 Oct 8 [PubMed PMID: 34625422]
Fabian ID, Rosser E, Sagoo MS. Epidemiological and genetic considerations in retinoblastoma. Community eye health. 2018:31(101):29-30 [PubMed PMID: 29915469]
Level 2 (mid-level) evidenceRosendahl Huber A, Van Hoeck A, Van Boxtel R. The Mutagenic Impact of Environmental Exposures in Human Cells and Cancer: Imprints Through Time. Frontiers in genetics. 2021:12():760039. doi: 10.3389/fgene.2021.760039. Epub 2021 Oct 20 [PubMed PMID: 34745228]
Rodolaki K, Pergialiotis V, Iakovidou N, Boutsikou T, Iliodromiti Z, Kanaka-Gantenbein C. The impact of maternal diabetes on the future health and neurodevelopment of the offspring: a review of the evidence. Frontiers in endocrinology. 2023:14():1125628. doi: 10.3389/fendo.2023.1125628. Epub 2023 Jul 3 [PubMed PMID: 37469977]
Athavale V, Khetan V. Knudson to embryo selection: A story of the genetics of retinoblastoma. Taiwan journal of ophthalmology. 2018 Oct-Dec:8(4):196-204. doi: 10.4103/tjo.tjo_37_18. Epub [PubMed PMID: 30637191]
Akdeniz Odemis D, Kebudi R, Bayramova J, Kilic Erciyas S, Kuru Turkcan G, Tuncer SB, Sukruoglu Erdogan O, Celik B, Kurt Gultaslar B, Buyukkapu Bay S, Tuncer S, Yazici H. RB1 gene mutations and genetic spectrum in retinoblastoma cases. Medicine. 2023 Sep 8:102(36):e35068. doi: 10.1097/MD.0000000000035068. Epub [PubMed PMID: 37682130]
Level 3 (low-level) evidenceDarwich R, Ghazawi FM, Rahme E, Alghazawi N, Burnier JV, Sasseville D, Burnier MN, Litvinov IV. Retinoblastoma Incidence Trends in Canada: A National Comprehensive Population-Based Study. Journal of pediatric ophthalmology and strabismus. 2019 Mar 19:56(2):124-130. doi: 10.3928/01913913-20190128-02. Epub [PubMed PMID: 30889267]
Aerts I, Lumbroso-Le Rouic L, Gauthier-Villars M, Brisse H, Doz F, Desjardins L. Retinoblastoma. Orphanet journal of rare diseases. 2006 Aug 25:1():31 [PubMed PMID: 16934146]
Liu Z, Yang Q, Cai N, Jin L, Zhang T, Chen X. Enigmatic Differences by Sex in Cancer Incidence: Evidence From Childhood Cancers. American journal of epidemiology. 2019 Jun 1:188(6):1130-1135. doi: 10.1093/aje/kwz058. Epub [PubMed PMID: 30834440]
Fabian ID, Khetan V, Stacey AW, Allen Foster, Ademola-Popoola DS, Berry JL, Cassoux N, Chantada GL, Hessissen L, Kaliki S, Kivelä TT, Luna-Fineman S, Munier FL, Reddy MA, Rojanaporn D, Blum S, Sherief ST, Staffieri SE, Theophile T, Waddell K, Ji X, Astbury NJ, Bascaran C, Burton M, Zondervan M, Bowman R, Global Retinoblastoma Study Group. Sex, gender, and retinoblastoma: analysis of 4351 patients from 153 countries. Eye (London, England). 2022 Aug:36(8):1571-1577. doi: 10.1038/s41433-021-01675-y. Epub 2021 Jul 16 [PubMed PMID: 34272514]
Schaiquevich P, Francis JH, Cancela MB, Carcaboso AM, Chantada GL, Abramson DH. Treatment of Retinoblastoma: What Is the Latest and What Is the Future. Frontiers in oncology. 2022:12():822330. doi: 10.3389/fonc.2022.822330. Epub 2022 Apr 1 [PubMed PMID: 35433448]
Cotache-Condor C, Kantety V, Grimm A, Williamson J, Landrum KR, Schroeder K, Staton C, Majaliwa E, Tang S, Rice HE, Smith ER. Determinants of delayed childhood cancer care in low- and middle-income countries: A systematic review. Pediatric blood & cancer. 2023 Mar:70(3):e30175. doi: 10.1002/pbc.30175. Epub 2022 Dec 29 [PubMed PMID: 36579761]
Level 1 (high-level) evidenceByroju VV, Nadukkandy AS, Cordani M, Kumar LD. Retinoblastoma: present scenario and future challenges. Cell communication and signaling : CCS. 2023 Sep 4:21(1):226. doi: 10.1186/s12964-023-01223-z. Epub 2023 Sep 4 [PubMed PMID: 37667345]
PDQ Pediatric Treatment Editorial Board. Retinoblastoma Treatment (PDQ®): Health Professional Version. PDQ Cancer Information Summaries. 2002:(): [PubMed PMID: 26389442]
Level 3 (low-level) evidenceGelband H, Jha P, Sankaranarayanan R, Horton S, Gupta S, Howard SC, Hunger SP, Antillon FG, Metzger ML, Israels T, Harif M, Rodriguez-Galindo C. Treating Childhood Cancer in Low- and Middle-Income Countries. Cancer: Disease Control Priorities, Third Edition (Volume 3). 2015 Nov 1:(): [PubMed PMID: 26913338]
Karaoui MS. Retinoblastoma: A new challenge to the Knudson's Dogma. Saudi journal of ophthalmology : official journal of the Saudi Ophthalmological Society. 2013 Jul:27(3):133-4. doi: 10.1016/j.sjopt.2013.07.008. Epub 2013 Jul 17 [PubMed PMID: 24227976]
Chen HZ, Tsai SY, Leone G. Emerging roles of E2Fs in cancer: an exit from cell cycle control. Nature reviews. Cancer. 2009 Nov:9(11):785-97. doi: 10.1038/nrc2696. Epub [PubMed PMID: 19851314]
Indovina P, Pentimalli F, Casini N, Vocca I, Giordano A. RB1 dual role in proliferation and apoptosis: cell fate control and implications for cancer therapy. Oncotarget. 2015 Jul 20:6(20):17873-90 [PubMed PMID: 26160835]
Lugano R, Ramachandran M, Dimberg A. Tumor angiogenesis: causes, consequences, challenges and opportunities. Cellular and molecular life sciences : CMLS. 2020 May:77(9):1745-1770. doi: 10.1007/s00018-019-03351-7. Epub 2019 Nov 6 [PubMed PMID: 31690961]
Dyer MA. Lessons from Retinoblastoma: Implications for Cancer, Development, Evolution, and Regenerative Medicine. Trends in molecular medicine. 2016 Oct:22(10):863-876. doi: 10.1016/j.molmed.2016.07.010. Epub 2016 Aug 23 [PubMed PMID: 27567287]
Grossniklaus HE. Retinoblastoma. Fifty years of progress. The LXXI Edward Jackson Memorial Lecture. American journal of ophthalmology. 2014 Nov:158(5):875-91. doi: 10.1016/j.ajo.2014.07.025. Epub 2014 Jul 24 [PubMed PMID: 25065496]
Kakarala CL, Raval VR, Mallu A, Rao R, Gavara S, Reddy VAP, Mishra DK, Jakati S, Kaliki S. Metastatic retinoblastoma at presentation: Clinical presentation, treatment, and outcomes. Oman journal of ophthalmology. 2023 Sep-Dec:16(3):524-528. doi: 10.4103/ojo.ojo_176_22. Epub 2023 Oct 18 [PubMed PMID: 38059077]
Kanukollu VM, Tripathy K. Leukocoria. StatPearls. 2022 Jan:(): [PubMed PMID: 32809629]
Lee C, Kim JK. Chromatin regulators in retinoblastoma: Biological roles and therapeutic applications. Journal of cellular physiology. 2021 Apr:236(4):2318-2332. doi: 10.1002/jcp.30022. Epub 2020 Aug 25 [PubMed PMID: 32840881]
Singh L, Kashyap S. Update on pathology of retinoblastoma. International journal of ophthalmology. 2018:11(12):2011-2016. doi: 10.18240/ijo.2018.12.22. Epub 2018 Dec 18 [PubMed PMID: 30588438]
Rajwanshi A, Srinivas R, Upasana G. Malignant small round cell tumors. Journal of cytology. 2009 Jan:26(1):1-10. doi: 10.4103/0970-9371.54861. Epub [PubMed PMID: 21938141]
Bogenmann E. Retinoblastoma cell differentiation in culture. International journal of cancer. 1986 Dec 15:38(6):883-7 [PubMed PMID: 3025105]
Chong EM, Coffee RE, Chintagumpala M, Hurwitz RL, Hurwitz MY, Chévez-Barrios P. Extensively necrotic retinoblastoma is associated with high-risk prognostic factors. Archives of pathology & laboratory medicine. 2006 Nov:130(11):1669-72 [PubMed PMID: 17076529]
Rössler J, Dietrich T, Pavlakovic H, Schweigerer L, Havers W, Schüler A, Bornfeld N, Schilling H. Higher vessel densities in retinoblastoma with local invasive growth and metastasis. The American journal of pathology. 2004 Feb:164(2):391-4 [PubMed PMID: 14742245]
Mendoza PR, Specht CS, Hubbard GB, Wells JR, Lynn MJ, Zhang Q, Kong J, Grossniklaus HE. Histopathologic grading of anaplasia in retinoblastoma. American journal of ophthalmology. 2015 Apr:159(4):764-76. doi: 10.1016/j.ajo.2014.12.014. Epub 2014 Dec 19 [PubMed PMID: 25528954]
Abusayf MM, Alkatan HM, Elkhamary S, Almesfer SA, Maktabi AMY. Histopathological assessment of optic nerve invasion guided by radiological findings in enucleated globes with retinoblastoma. BMC ophthalmology. 2020 Sep 29:20(1):386. doi: 10.1186/s12886-020-01654-z. Epub 2020 Sep 29 [PubMed PMID: 32993566]
Abramson DH, Gobin YP, Dunkel IJ, Francis JH. Successful Treatment of Massive Choroidal Invasion in Retinoblastoma with Intra-arterial Chemotherapy (Ophthalmic Artery Chemosurgery). Ophthalmology. Retina. 2021 Sep:5(9):936-939. doi: 10.1016/j.oret.2020.12.018. Epub 2020 Dec 29 [PubMed PMID: 33383259]
Kajal S, Cui D, Winters R. Malignant Orbital Tumors. StatPearls. 2025 Jan:(): [PubMed PMID: 38753918]
Munier FL. Classification and management of seeds in retinoblastoma. Ellsworth Lecture Ghent August 24th 2013. Ophthalmic genetics. 2014 Dec:35(4):193-207. doi: 10.3109/13816810.2014.973045. Epub 2014 Oct 16 [PubMed PMID: 25321846]
Rodrigues MM, Wiggert B, Shields J, Donoso L, Bardenstein D, Katz N, Friendly D, Chader G. Retinoblastoma. Immunohistochemistry and cell differentiation. Ophthalmology. 1987 Apr:94(4):378-87 [PubMed PMID: 3495765]
Rao AA, Naheedy JH, Chen JY, Robbins SL, Ramkumar HL. A clinical update and radiologic review of pediatric orbital and ocular tumors. Journal of oncology. 2013:2013():975908. doi: 10.1155/2013/975908. Epub 2013 Mar 12 [PubMed PMID: 23577029]
Fabian ID, Reddy A, Sagoo MS. Classification and staging of retinoblastoma. Community eye health. 2018:31(101):11-13 [PubMed PMID: 29915461]
Dixit R, Riviere J, Krishnan K, Andersen ME. Toxicokinetics and physiologically based toxicokinetics in toxicology and risk assessment. Journal of toxicology and environmental health. Part B, Critical reviews. 2003 Jan-Feb:6(1):1-40 [PubMed PMID: 12587252]
Yanık Ö, Gündüz K, Yavuz K, Taçyıldız N, Ünal E. Chemotherapy in Retinoblastoma: Current Approaches. Turkish journal of ophthalmology. 2015 Dec:45(6):259-267 [PubMed PMID: 27800245]
Kim HJ, Di Nicola M, Augsburger JJ, Williams BK. Successful salvage intravenous chemotherapy after tandem selective ophthalmic artery infusion chemotherapy in bilateral retinoblastoma. Indian journal of ophthalmology. 2020 Nov:68(11):2618-2620. doi: 10.4103/ijo.IJO_576_20. Epub [PubMed PMID: 33120714]
Markowicz-Piasecka M, Markiewicz A, Darłak P, Sikora J, Adla SK, Bagina S, Huttunen KM. Current Chemical, Biological, and Physiological Views in the Development of Successful Brain-Targeted Pharmaceutics. Neurotherapeutics : the journal of the American Society for Experimental NeuroTherapeutics. 2022 Apr:19(3):942-976. doi: 10.1007/s13311-022-01228-5. Epub 2022 Apr 7 [PubMed PMID: 35391662]
Dennison JB, Jones DR, Renbarger JL, Hall SD. Effect of CYP3A5 expression on vincristine metabolism with human liver microsomes. The Journal of pharmacology and experimental therapeutics. 2007 May:321(2):553-63 [PubMed PMID: 17272675]
Beumer JH, Inker LA, Levey AS. Improving Carboplatin Dosing Based on Estimated GFR. American journal of kidney diseases : the official journal of the National Kidney Foundation. 2018 Feb:71(2):163-165. doi: 10.1053/j.ajkd.2017.10.005. Epub 2017 Dec 6 [PubMed PMID: 29217306]
Tan YM, Barton HA, Boobis A, Brunner R, Clewell H, Cope R, Dawson J, Domoradzki J, Egeghy P, Gulati P, Ingle B, Kleinstreuer N, Lowe K, Lowit A, Mendez E, Miller D, Minucci J, Nguyen J, Paini A, Perron M, Phillips K, Qian H, Ramanarayanan T, Sewell F, Villanueva P, Wambaugh J, Embry M. Opportunities and challenges related to saturation of toxicokinetic processes: Implications for risk assessment. Regulatory toxicology and pharmacology : RTP. 2021 Dec:127():105070. doi: 10.1016/j.yrtph.2021.105070. Epub 2021 Oct 28 [PubMed PMID: 34718074]
Level 2 (mid-level) evidenceLegha SS. Vincristine neurotoxicity. Pathophysiology and management. Medical toxicology. 1986 Nov-Dec:1(6):421-7 [PubMed PMID: 3540519]
Cheng YJ, Wu R, Cheng ML, Du J, Hu XW, Yu L, Zhao XK, Yao YM, Long QZ, Zhu LL, Zhu JJ, Huang NW, Liu HJ, Hu YX, Wan F. Carboplatin-induced hematotoxicity among patients with non-small cell lung cancer: Analysis on clinical adverse events and drug-gene interactions. Oncotarget. 2017 May 9:8(19):32228-32236. doi: 10.18632/oncotarget.12951. Epub [PubMed PMID: 27802181]
Zhang W, Gou P, Dupret JM, Chomienne C, Rodrigues-Lima F. Etoposide, an anticancer drug involved in therapy-related secondary leukemia: Enzymes at play. Translational oncology. 2021 Oct:14(10):101169. doi: 10.1016/j.tranon.2021.101169. Epub 2021 Jul 6 [PubMed PMID: 34243013]
Francis JH, Schaiquevich P, Buitrago E, Del Sole MJ, Zapata G, Croxatto JO, Marr BP, Brodie SE, Berra A, Chantada GL, Abramson DH. Local and systemic toxicity of intravitreal melphalan for vitreous seeding in retinoblastoma: a preclinical and clinical study. Ophthalmology. 2014 Sep:121(9):1810-7. doi: 10.1016/j.ophtha.2014.03.028. Epub 2014 May 10 [PubMed PMID: 24819859]
O'Reilly S, Rowinsky EK, Slichenmyer W, Donehower RC, Forastiere AA, Ettinger DS, Chen TL, Sartorius S, Grochow LB. Phase I and pharmacologic study of topotecan in patients with impaired renal function. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 1996 Dec:14(12):3062-73 [PubMed PMID: 8955651]
Kaestner SA, Sewell GJ. Chemotherapy dosing part I: scientific basis for current practice and use of body surface area. Clinical oncology (Royal College of Radiologists (Great Britain)). 2007 Feb:19(1):23-37 [PubMed PMID: 17305252]
Manjandavida FP, Stathopoulos C, Zhang J, Honavar SG, Shields CL. Intra-arterial chemotherapy in retinoblastoma - A paradigm change. Indian journal of ophthalmology. 2019 Jun:67(6):740-754. doi: 10.4103/ijo.IJO_866_19. Epub [PubMed PMID: 31124482]
Bayat Mokhtari R, Homayouni TS, Baluch N, Morgatskaya E, Kumar S, Das B, Yeger H. Combination therapy in combating cancer. Oncotarget. 2017 Jun 6:8(23):38022-38043. doi: 10.18632/oncotarget.16723. Epub [PubMed PMID: 28410237]
Jansen RW,de Bloeme CM,Brisse HJ,Galluzzi P,Cardoen L,Göricke S,Maeder P,Cassoux N,Gauthier A,Schlueter S,Hadjistilianou T,Munier FL,Castelijns JA,van der Valk P,Moll AC,de Jong MC,de Graaf P, MR Imaging Features to Differentiate Retinoblastoma from Coats' Disease and Persistent Fetal Vasculature. Cancers. 2020 Nov 30 [PubMed PMID: 33266342]
Das A, Thulaseedharan DK, Shah PK, Subramaniam P, Venkatapathy N. Spectrum and demographic profile of pediatric intraocular tumors from a single center in India: an analysis of 445 eyes. Graefe's archive for clinical and experimental ophthalmology = Albrecht von Graefes Archiv fur klinische und experimentelle Ophthalmologie. 2024 Dec 30:():. doi: 10.1007/s00417-024-06728-y. Epub 2024 Dec 30 [PubMed PMID: 39739100]
Kang E, Suk-Gyu H. Changes in PAX7 Positive Satellite Cells in Extraocular Muscle After Strabismus Surgery. Journal of pediatric ophthalmology and strabismus. 2025 Jan 21:():1-6. doi: 10.3928/01913913-20241210-02. Epub 2025 Jan 21 [PubMed PMID: 39835589]
Gupta P, Gurnani B, Patel BC. Pediatric Cataract. StatPearls. 2025 Jan:(): [PubMed PMID: 34283446]
Kaliki S, Ji X, Zou Y, Rashid R, Sultana S, Taju Sherief S, Cassoux N, Y Diaz Coronado R, Luis Garcia Leon J, López AMZ, Polyakov VG, Ushakova TL, Rani Roy S, Ahmad A, Al Harby L, Reddy MA, Sagoo MS, L Berry J, Kim J, Polski A, Astbury NJ, Bascaran C, Blum S, Bowman R, Burton MJ, Foster A, Gomel N, Keren-Froim N, Madgar S, W Stacey A, M Steinberg D, Mohamed A, Zondervan M, Fabian ID. Lag Time between Onset of First Symptom and Treatment of Retinoblastoma: An International Collaborative Study of 692 Patients from 10 Countries. Cancers. 2021 Apr 19:13(8):. doi: 10.3390/cancers13081956. Epub 2021 Apr 19 [PubMed PMID: 33921566]
Pinto VL, Tadi P, Adeyinka A. Increased Intracranial Pressure. StatPearls. 2024 Jan:(): [PubMed PMID: 29489250]
Marković L, Bukovac A, Varošanec AM, Šlaus N, Pećina-Šlaus N. Genetics in ophthalmology: molecular blueprints of retinoblastoma. Human genomics. 2023 Sep 1:17(1):82. doi: 10.1186/s40246-023-00529-w. Epub 2023 Sep 1 [PubMed PMID: 37658463]
Ergaz Z, Ornoy A. Perinatal and early postnatal factors underlying developmental delay and disabilities. Developmental disabilities research reviews. 2011:17(2):59-70. doi: 10.1002/ddrr.1101. Epub [PubMed PMID: 23362026]
Choe SA, Eliot MN, Savitz DA, Wellenius GA. Ambient air pollution during pregnancy and risk of gestational diabetes in New York City. Environmental research. 2019 Aug:175():414-420. doi: 10.1016/j.envres.2019.04.030. Epub 2019 May 24 [PubMed PMID: 31154231]
Hummell AC, Cummings M. Role of the nutrition-focused physical examination in identifying malnutrition and its effectiveness. Nutrition in clinical practice : official publication of the American Society for Parenteral and Enteral Nutrition. 2022 Feb:37(1):41-49. doi: 10.1002/ncp.10797. Epub 2021 Nov 9 [PubMed PMID: 34751967]
Ronquillo Y, Zeppieri M, Patel BC. Nonspecific Orbital Inflammation. StatPearls. 2025 Jan:(): [PubMed PMID: 31869057]
. How to test for the red reflex in a child. Community eye health. 2014:27(86):36 [PubMed PMID: 25316966]
Hull S, Tailor V, Balduzzi S, Rahi J, Schmucker C, Virgili G, Dahlmann-Noor A. Tests for detecting strabismus in children aged 1 to 6 years in the community. The Cochrane database of systematic reviews. 2017 Nov 6:11(11):CD011221. doi: 10.1002/14651858.CD011221.pub2. Epub 2017 Nov 6 [PubMed PMID: 29105728]
Level 1 (high-level) evidenceChristy J, Jain N, Gurnani B, Kaur K. Twinkling Eye -A Rare Presentation in Neovascular Glaucoma. Journal of glaucoma. 2019 May 23:():. doi: 10.1097/IJG.0000000000001287. Epub 2019 May 23 [PubMed PMID: 31135586]
Walker HK, Hall WD, Hurst JW, Schneiderman H. The Funduscopic Examination. Clinical Methods: The History, Physical, and Laboratory Examinations. 1990:(): [PubMed PMID: 21250065]
Friedman DI. Papilledema and idiopathic intracranial hypertension. Continuum (Minneapolis, Minn.). 2014 Aug:20(4 Neuro-ophthalmology):857-76. doi: 10.1212/01.CON.0000453314.75261.66. Epub [PubMed PMID: 25099098]
López F, Rodrigo JP, Silver CE, Haigentz M Jr, Bishop JA, Strojan P, Hartl DM, Bradley PJ, Mendenhall WM, Suárez C, Takes RP, Hamoir M, Robbins KT, Shaha AR, Werner JA, Rinaldo A, Ferlito A. Cervical lymph node metastases from remote primary tumor sites. Head & neck. 2016 Apr:38 Suppl 1(Suppl 1):E2374-85. doi: 10.1002/hed.24344. Epub 2015 Dec 29 [PubMed PMID: 26713674]
Feroze KB, Blair K, Patel BC. Buphthalmos. StatPearls. 2025 Jan:(): [PubMed PMID: 28613637]
Balmer A, Munier F. Differential diagnosis of leukocoria and strabismus, first presenting signs of retinoblastoma. Clinical ophthalmology (Auckland, N.Z.). 2007 Dec:1(4):431-9 [PubMed PMID: 19668520]
Shafiq A. Seeing red in young children: the importance of the red reflex. The British journal of general practice : the journal of the Royal College of General Practitioners. 2015 Apr:65(633):209-10. doi: 10.3399/bjgp15X684625. Epub [PubMed PMID: 25824175]
Dhillon HK, Agarkar S, Vijaya L, Bhende M, Baskaran M, Jaichandran VV. Examination under anesthesia: Preferred Practice. Indian journal of ophthalmology. 2023 Nov:71(11):3438-3445. doi: 10.4103/IJO.IJO_3259_22. Epub [PubMed PMID: 37870002]
De La Hoz Polo M, Torramilans Lluís A, Pozuelo Segura O, Anguera Bosque A, Esmerado Appiani C, Caminal Mitjana JM. Ocular ultrasonography focused on the posterior eye segment: what radiologists should know. Insights into imaging. 2016 Jun:7(3):351-64. doi: 10.1007/s13244-016-0471-z. Epub 2016 Feb 24 [PubMed PMID: 26910565]
de Graaf P, Göricke S, Rodjan F, Galluzzi P, Maeder P, Castelijns JA, Brisse HJ, European Retinoblastoma Imaging Collaboration (ERIC). Guidelines for imaging retinoblastoma: imaging principles and MRI standardization. Pediatric radiology. 2012 Jan:42(1):2-14. doi: 10.1007/s00247-011-2201-5. Epub 2011 Aug 18 [PubMed PMID: 21850471]
Florkow MC, Willemsen K, Mascarenhas VV, Oei EHG, van Stralen M, Seevinck PR. Magnetic Resonance Imaging Versus Computed Tomography for Three-Dimensional Bone Imaging of Musculoskeletal Pathologies: A Review. Journal of magnetic resonance imaging : JMRI. 2022 Jul:56(1):11-34. doi: 10.1002/jmri.28067. Epub 2022 Jan 19 [PubMed PMID: 35044717]
Adhi M, Duker JS. Optical coherence tomography--current and future applications. Current opinion in ophthalmology. 2013 May:24(3):213-21. doi: 10.1097/ICU.0b013e32835f8bf8. Epub [PubMed PMID: 23429598]
Level 3 (low-level) evidenceFakeri M, Shakoul F, Yaghoubi SM, Koulaeizadeh S, Haghi M. Comprehensive insights into circular RNAs, miRNAs, and lncRNAs as biomarkers in retinoblastoma. Ophthalmic genetics. 2025 Jan 23:():1-11. doi: 10.1080/13816810.2025.2456607. Epub 2025 Jan 23 [PubMed PMID: 39849678]
Tomar AS, Finger PT, Gallie B, Kivelä TT, Mallipatna A, Zhang C, Zhao J, Wilson MW, Brennan RC, Burges M, Kim J, Berry JL, Jubran R, Khetan V, Ganesan S, Yarovoy A, Yarovaya V, Kotova E, Volodin D, Yousef YA, Nummi K, Ushakova TL, Yugay OV, Polyakov VG, Ramirez-Ortiz MA, Esparza-Aguiar E, Chantada G, Schaiquevich P, Fandino A, Yam JC, Lau WW, Lam CP, Sharwood P, Moorthy S, Long QB, Essuman VA, Renner LA, Semenova E, Català-Mora J, Correa-Llano G, Carreras E, American Joint Committee on Cancer Ophthalmic Oncology Task Force. High-risk Pathologic Features Based on Presenting Findings in Advanced Intraocular Retinoblastoma: A Multicenter, International Data-Sharing American Joint Committee on Cancer Study. Ophthalmology. 2022 Aug:129(8):923-932. doi: 10.1016/j.ophtha.2022.04.006. Epub 2022 Apr 15 [PubMed PMID: 35436535]
Das D, Bhattacharjee K, Barthakur SS, Tahiliani PS, Deka P, Bhattacharjee H, Deka A, Paul R. A new rosette in retinoblastoma. Indian journal of ophthalmology. 2014 May:62(5):638-41. doi: 10.4103/0301-4738.129786. Epub [PubMed PMID: 24881618]
Mjønes P, Sagatun L, Nordrum IS, Waldum HL. Neuron-Specific Enolase as an Immunohistochemical Marker Is Better Than Its Reputation. The journal of histochemistry and cytochemistry : official journal of the Histochemistry Society. 2017 Dec:65(12):687-703. doi: 10.1369/0022155417733676. Epub 2017 Oct 3 [PubMed PMID: 28972818]
Rosen RD, Sapra A. TNM Classification. StatPearls. 2025 Jan:(): [PubMed PMID: 31985980]
Rindy LJ,Chambers AR, Bone Marrow Aspiration And Biopsy. StatPearls. 2022 Jan [PubMed PMID: 32644658]
Maitray A, Khetan V. Classification of retinoblastoma: Evolution with time and the need for uniformity. Oman journal of ophthalmology. 2017 Sep-Dec:10(3):133-134. doi: 10.4103/0974-620X.216017. Epub [PubMed PMID: 29118485]
Ancona-Lezama D,Dalvin LA,Shields CL, Modern treatment of retinoblastoma: A 2020 review. Indian journal of ophthalmology. 2020 Nov [PubMed PMID: 33120616]
Mathus-Vliegen EM, Tytgat GN. Laser photocoagulation in the palliation of colorectal malignancies. Cancer. 1986 Jun 1:57(11):2212-6 [PubMed PMID: 2421867]
Shields JA, Parsons H, Shields CL, Giblin ME. The role of cryotherapy in the management of retinoblastoma. American journal of ophthalmology. 1989 Sep 15:108(3):260-4 [PubMed PMID: 2774035]
Thomas GN, Chou IL, Gopal L. Plaque Radiotherapy for Ocular Melanoma. Cancers. 2024 Oct 3:16(19):. doi: 10.3390/cancers16193386. Epub 2024 Oct 3 [PubMed PMID: 39410006]
Romanowska-Dixon B, Kowal J, Pogrzebielski A, Markiewicz A. [Transpupillary thermotherapy (TTT) for intraocular metastases in choroid]. Klinika oczna. 2011:113(4-6):132-5 [PubMed PMID: 21913441]
Fabian ID, Johnson KP, Stacey AW, Sagoo MS, Reddy MA. Focal laser treatment in addition to chemotherapy for retinoblastoma. The Cochrane database of systematic reviews. 2017 Jun 7:6(6):CD012366. doi: 10.1002/14651858.CD012366.pub2. Epub 2017 Jun 7 [PubMed PMID: 28589646]
Level 1 (high-level) evidenceGatzemeier U, von Pawel J, Laumen R, Hossfeld DK, Neuhauss R, Reck M, Lenaz L. Carboplatin/etoposide/vincristine therapy in small cell lung cancer. Oncology. 1992:49 Suppl 1():25-33 [PubMed PMID: 1323809]
Yousef YA, Noureldin AM, Sultan I, Deebajah R, Al-Hussaini M, Shawagfeh M, Mehyar M, Mohammad M, Jaradat I, AlNawaiseh I. Intravitreal Melphalan Chemotherapy for Vitreous Seeds in Retinoblastoma. Journal of ophthalmology. 2020:2020():8628525. doi: 10.1155/2020/8628525. Epub 2020 Jan 24 [PubMed PMID: 32047663]
Schefler AC, Kim RS. Recent advancements in the management of retinoblastoma and uveal melanoma. F1000Research. 2018:7():. pii: F1000 Faculty Rev-476. doi: 10.12688/f1000research.11941.1. Epub 2018 Apr 18 [PubMed PMID: 29755733]
Pelton RW, Patel BC. Superomedial lid crease approach to the medial intraconal space: a new technique for access to the optic nerve and central space. Ophthalmic plastic and reconstructive surgery. 2001 Jul:17(4):241-53 [PubMed PMID: 11476174]
Level 3 (low-level) evidenceFeng ZX, Zhao J, Zhang N, Jin M, Gallie B. Adjuvant Chemotherapy Improves Survival for Children With Massive Choroidal Invasion of Retinoblastoma. Investigative ophthalmology & visual science. 2023 Aug 1:64(11):27. doi: 10.1167/iovs.64.11.27. Epub [PubMed PMID: 37603354]
Elaraoud I, Ch'ng S, Karl D, Kalogeropoulos D, Chavan R, Sharma A. Management of retinal detachment in retinoblastoma with globe conserving treatment. Journal of current ophthalmology. 2019 Mar:31(1):43-48. doi: 10.1016/j.joco.2018.09.002. Epub 2018 Sep 22 [PubMed PMID: 30899845]
Bagley AF, Grosshans DR, Philip NV, Foster J, McAleer MF, McGovern SL, Lassen-Ramshad Y, Mahajan A, Paulino AC. Efficacy of proton therapy in children with high-risk and locally recurrent neuroblastoma. Pediatric blood & cancer. 2019 Aug:66(8):e27786. doi: 10.1002/pbc.27786. Epub 2019 May 2 [PubMed PMID: 31050179]
Kamihara J, Bourdeaut F, Foulkes WD, Molenaar JJ, Mossé YP, Nakagawara A, Parareda A, Scollon SR, Schneider KW, Skalet AH, States LJ, Walsh MF, Diller LR, Brodeur GM. Retinoblastoma and Neuroblastoma Predisposition and Surveillance. Clinical cancer research : an official journal of the American Association for Cancer Research. 2017 Jul 1:23(13):e98-e106. doi: 10.1158/1078-0432.CCR-17-0652. Epub [PubMed PMID: 28674118]
Knudsen ES, Wang JY. Targeting the RB-pathway in cancer therapy. Clinical cancer research : an official journal of the American Association for Cancer Research. 2010 Feb 15:16(4):1094-9. doi: 10.1158/1078-0432.CCR-09-0787. Epub 2010 Feb 9 [PubMed PMID: 20145169]
Daniels AB, Sishtla KL, Bogan CM, Pierce JM, Chen SC, Xu L, Berry JL, Corson TW. Aqueous VEGF-A Levels as a Liquid Biopsy Biomarker of Retinoblastoma Vitreous Seed Response to Therapy. Investigative ophthalmology & visual science. 2024 Jun 3:65(6):18. doi: 10.1167/iovs.65.6.18. Epub [PubMed PMID: 38861274]
Mehta A, Singh SR, Dogra M, Ram J. Persistent fetal vasculature - Clinical spectrum. Indian journal of ophthalmology. 2018 Dec:66(12):1860. doi: 10.4103/ijo.IJO_1042_18. Epub [PubMed PMID: 30451201]
Shields JA, Shields CL, Honavar SG, Demirci H. Clinical variations and complications of Coats disease in 150 cases: the 2000 Sanford Gifford Memorial Lecture. American journal of ophthalmology. 2001 May:131(5):561-71 [PubMed PMID: 11336930]
Level 2 (mid-level) evidenceAhn SJ, Ryoo NK, Woo SJ. Ocular toxocariasis: clinical features, diagnosis, treatment, and prevention. Asia Pacific allergy. 2014 Jul:4(3):134-41. doi: 10.5415/apallergy.2014.4.3.134. Epub 2014 Jul 29 [PubMed PMID: 25097848]
Wheatley CM, Dickinson JL, Mackey DA, Craig JE, Sale MM. Retinopathy of prematurity: recent advances in our understanding. The British journal of ophthalmology. 2002 Jun:86(6):696-700 [PubMed PMID: 12034695]
Level 3 (low-level) evidenceTauqeer Z, Yonekawa Y. Familial Exudative Vitreoretinopathy: Pathophysiology, Diagnosis, and Management. Asia-Pacific journal of ophthalmology (Philadelphia, Pa.). 2018 May-Jun:7(3):176-182. doi: 10.22608/APO.201855. Epub 2018 Apr 9 [PubMed PMID: 29633588]
Tadepalli SH, Shields CL, Shields JA, Honavar SG. Intraocular medulloepithelioma - A review of clinical features, DICER 1 mutation, and management. Indian journal of ophthalmology. 2019 Jun:67(6):755-762. doi: 10.4103/ijo.IJO_845_19. Epub [PubMed PMID: 31124483]
Martin K, Rossi V, Ferrucci S, Pian D. Retinal astrocytic hamartoma. Optometry (St. Louis, Mo.). 2010 May:81(5):221-33. doi: 10.1016/j.optm.2009.12.009. Epub [PubMed PMID: 20435268]
Adam MP, Feldman J, Mirzaa GM, Pagon RA, Wallace SE, Amemiya A, Scruggs BA, Reding MQ, Schimmenti LA. NDP-Related Retinopathies. GeneReviews(®). 1993:(): [PubMed PMID: 20301506]
Lingam G, Sen AC, Lingam V, Bhende M, Padhi TR, Xinyi S. Ocular coloboma-a comprehensive review for the clinician. Eye (London, England). 2021 Aug:35(8):2086-2109. doi: 10.1038/s41433-021-01501-5. Epub 2021 Mar 21 [PubMed PMID: 33746210]
Jena S, Tripathy K. Vitreous Hemorrhage. StatPearls. 2025 Jan:(): [PubMed PMID: 32644557]
Mahabadi N, Gurnani B, Czyz CN. Bacterial Endophthalmitis. StatPearls. 2025 Jan:(): [PubMed PMID: 31424768]
Lee RW, Nicholson LB, Sen HN, Chan CC, Wei L, Nussenblatt RB, Dick AD. Autoimmune and autoinflammatory mechanisms in uveitis. Seminars in immunopathology. 2014 Sep:36(5):581-94. doi: 10.1007/s00281-014-0433-9. Epub 2014 May 24 [PubMed PMID: 24858699]
Ye H, Xue K, Zhang P, Chen R, Zhai X, Ling L, Xiao W, Tang L, Wang H, Mao Y, Ai S, Bi Y, Liu Q, Zou Y, Qian J, Yang H. Three vs 6 Cycles of Chemotherapy for High-Risk Retinoblastoma: A Randomized Clinical Trial. JAMA. 2024 Nov 19:332(19):1634-1641. doi: 10.1001/jama.2024.19981. Epub [PubMed PMID: 39432296]
Level 1 (high-level) evidenceYousef YA, Al Jboor M, Mohammad M, Mehyar M, Toro MD, Nazzal R, Alzureikat QH, Rejdak M, Elfalah M, Sultan I, Rejdak R, Al-Hussaini M, Al-Nawaiseh I. Safety and Efficacy of Intravitreal Chemotherapy (Melphalan) to Treat Vitreous Seeds in Retinoblastoma. Frontiers in pharmacology. 2021:12():696787. doi: 10.3389/fphar.2021.696787. Epub 2021 Jul 12 [PubMed PMID: 34322023]
Dimaras H, Corson TW, Cobrinik D, White A, Zhao J, Munier FL, Abramson DH, Shields CL, Chantada GL, Njuguna F, Gallie BL. Retinoblastoma. Nature reviews. Disease primers. 2015 Aug 27:1():15021. doi: 10.1038/nrdp.2015.21. Epub 2015 Aug 27 [PubMed PMID: 27189421]
Pareek A, Kumar D, Pareek A, Gupta MM, Jeandet P, Ratan Y, Jain V, Kamal MA, Saboor M, Ashraf GM, Chuturgoon A. Retinoblastoma: An update on genetic origin, classification, conventional to next-generation treatment strategies. Heliyon. 2024 Jun 30:10(12):e32844. doi: 10.1016/j.heliyon.2024.e32844. Epub 2024 Jun 11 [PubMed PMID: 38975183]
Lumbroso-Le Rouic L, Aerts I, Hajage D, Lévy-Gabriel C, Savignoni A, Algret N, Cassoux N, Bertozzi AI, Esteve M, Doz F, Desjardins L. Conservative treatment of retinoblastoma: a prospective phase II randomized trial of neoadjuvant chemotherapy followed by local treatments and chemothermotherapy. Eye (London, England). 2016 Jan:30(1):46-52. doi: 10.1038/eye.2015.179. Epub 2015 Oct 2 [PubMed PMID: 26427984]
Level 1 (high-level) evidenceTanveer S, Zafar F, Bibi H, Haroon H, Ahmad O, Iqbal MS, Zakir Z, Khilji M, Tanveer S, Hassan RE. Advancements in Retinoblastoma Treatment: Unraveling the Potential of Intravitreal Chemotherapy. Cureus. 2024 Jan:16(1):e53012. doi: 10.7759/cureus.53012. Epub 2024 Jan 26 [PubMed PMID: 38410326]
Peña-Balderas AM, Martínez-Sánchez M, Olmos-Sánchez I, Calderón-González K, Moctezuma-Dávila M, Rangel-Charqueño M, Hernández-Monge J, Olivares-Illana V. Analysis of pathogenic variants in retinoblastoma reveals a potential gain of function mutation. Genes & cancer. 2025:16():1-15. doi: 10.18632/genesandcancer.239. Epub 2025 Jan 20 [PubMed PMID: 39845333]
Silvera VM, Guerin JB, Brinjikji W, Dalvin LA. Retinoblastoma: What the Neuroradiologist Needs to Know. AJNR. American journal of neuroradiology. 2021 Apr:42(4):618-626. doi: 10.3174/ajnr.A6949. Epub 2021 Jan 28 [PubMed PMID: 33509920]
Chawla B, Jain A, Azad R. Conservative treatment modalities in retinoblastoma. Indian journal of ophthalmology. 2013 Sep:61(9):479-85. doi: 10.4103/0301-4738.119424. Epub [PubMed PMID: 24104705]
Teke Kısa P, Emir S. Role of surveillance screening in detecting tumor recurrence after treatment of childhood cancers. Turkish archives of pediatrics. 2021 Mar:56(2):147-151. doi: 10.14744/TurkPediatriArs.2020.38243. Epub 2021 Jan 6 [PubMed PMID: 34286325]
Labardini CP, Blumenthal EZ. Causative Pathogens in Endophthalmitis after Intravitreal Injection of Anti-vascular Endothelial Growth Factor Agents. Rambam Maimonides medical journal. 2018 Sep 2:9(4):1-6. doi: 10.5041/RMMJ.10348. Epub 2018 Sep 2 [PubMed PMID: 30180932]
Dracham CB, Shankar A, Madan R. Radiation induced secondary malignancies: a review article. Radiation oncology journal. 2018 Jun:36(2):85-94. doi: 10.3857/roj.2018.00290. Epub 2018 Jun 29 [PubMed PMID: 29983028]
Mularska W, Chicheł A, Rospond-Kubiak I. Radiation retinopathy following episcleral brachytherapy for intraocular tumors: Current treatment options. Journal of contemporary brachytherapy. 2023 Oct:15(5):372-382. doi: 10.5114/jcb.2023.132398. Epub 2023 Oct 26 [PubMed PMID: 38026080]
Fay A, Nallasamy N, Pemberton JD, Callahan A, Wladis EJ, Nguyen J, Durand ML, New England Oculoplastics Society Study Group. Prophylactic postoperative antibiotics for enucleation and evisceration. Ophthalmic plastic and reconstructive surgery. 2013 Jul-Aug:29(4):281-5. doi: 10.1097/IOP.0b013e3182916674. Epub [PubMed PMID: 23645357]
Dommering CJ, Marees T, van der Hout AH, Imhof SM, Meijers-Heijboer H, Ringens PJ, van Leeuwen FE, Moll AC. RB1 mutations and second primary malignancies after hereditary retinoblastoma. Familial cancer. 2012 Jun:11(2):225-33. doi: 10.1007/s10689-011-9505-3. Epub [PubMed PMID: 22205104]
Lee SY, Gurnani B, Mesfin FB. Blindness. StatPearls. 2025 Jan:(): [PubMed PMID: 28846303]
Glaser SLC, Fraterman I, van Brummelen N, Tibollo V, Del Campo LM, Mallo H, Wilgenhof S, Wilk S, Gisko V, Khadakou V, Cornet R, Ottaviano M, Medlock S. Usability and Usefulness of a Symptom Management Coaching System for Patients With Cancer Treated With Immune Checkpoint Inhibitors: Comparative Mixed Methods Study. JMIR formative research. 2025 Jan 23:9():e57659. doi: 10.2196/57659. Epub 2025 Jan 23 [PubMed PMID: 39847771]
Level 2 (mid-level) evidenceKan Z, Wen J, Bonato V, Webster J, Yang W, Ivanov V, Kim KH, Roh W, Liu C, Mu XJ, Lapira-Miller J, Oyer J, VanArsdale T, Rejto PA, Bienkowska J. Real-world clinical multi-omics analyses reveal bifurcation of ER-independent and ER-dependent drug resistance to CDK4/6 inhibitors. Nature communications. 2025 Jan 22:16(1):932. doi: 10.1038/s41467-025-55914-x. Epub 2025 Jan 22 [PubMed PMID: 39843429]
Mohammad M, Mehyar M, Halalsheh H, Shehada R, Al Adawi O, Khzouz J, Jaradat I, Al-Hussaini M, Sultan I, Alnawaiseh I, Yousef YA. The Impact of Tumor Laterality (Unilateral vs. Bilateral) on Presentation and Management Outcome in Patients with Retinoblastoma. Journal of clinical medicine. 2024 Apr 8:13(7):. doi: 10.3390/jcm13072146. Epub 2024 Apr 8 [PubMed PMID: 38610910]
Dare AJ, Knapp GC, Romanoff A, Olasehinde O, Famurewa OC, Komolafe AO, Olatoke S, Katung A, Alatise OI, Kingham TP. High-burden Cancers in Middle-income Countries: A Review of Prevention and Early Detection Strategies Targeting At-risk Populations. Cancer prevention research (Philadelphia, Pa.). 2021 Dec:14(12):1061-1074. doi: 10.1158/1940-6207.CAPR-20-0571. Epub 2021 Sep 10 [PubMed PMID: 34507972]
Warda O, Naeem Z, Roelofs KA, Sagoo MS, Reddy MA. Retinoblastoma and vision. Eye (London, England). 2023 Apr:37(5):797-808. doi: 10.1038/s41433-021-01845-y. Epub 2022 Jan 5 [PubMed PMID: 34987197]
Wong JR, Morton LM, Tucker MA, Abramson DH, Seddon JM, Sampson JN, Kleinerman RA. Risk of subsequent malignant neoplasms in long-term hereditary retinoblastoma survivors after chemotherapy and radiotherapy. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2014 Oct 10:32(29):3284-90. doi: 10.1200/JCO.2013.54.7844. Epub 2014 Sep 2 [PubMed PMID: 25185089]
Banerjee SC, Pottenger E, Petriccione M, Chou JF, Ford JS, Sklar CA, Robison LL, Kleinerman RA, Oeffinger KC, Francis JH, Abramson DH, Dunkel IJ, Friedman DN. Impact of enucleation on adult retinoblastoma survivors' quality of life: A qualitative study of survivors' perspectives. Palliative & supportive care. 2020 Jun:18(3):322-331. doi: 10.1017/S1478951519000920. Epub [PubMed PMID: 31699178]
Level 2 (mid-level) evidenceOrzetti S, Tommasi F, Bertola A, Bortolin G, Caccin E, Cecco S, Ferrarin E, Giacomin E, Baldo P. Genetic Therapy and Molecular Targeted Therapy in Oncology: Safety, Pharmacovigilance, and Perspectives for Research and Clinical Practice. International journal of molecular sciences. 2022 Mar 10:23(6):. doi: 10.3390/ijms23063012. Epub 2022 Mar 10 [PubMed PMID: 35328435]
Level 3 (low-level) evidencePalmisciano P, Ferini G, Ogasawara C, Wahood W, Bin Alamer O, Gupta AD, Scalia G, Larsen AMG, Yu K, Umana GE, Cohen-Gadol AA, El Ahmadieh TY, Haider AS. Orbital Metastases: A Systematic Review of Clinical Characteristics, Management Strategies, and Treatment Outcomes. Cancers. 2021 Dec 24:14(1):. doi: 10.3390/cancers14010094. Epub 2021 Dec 24 [PubMed PMID: 35008259]
Level 1 (high-level) evidenceJayarangaiah A, Kemp AK, Theetha Kariyanna P. Bone Metastasis. StatPearls. 2024 Jan:(): [PubMed PMID: 29939688]
Crawford J, Herndon D, Gmitter K, Weiss J. The impact of myelosuppression on quality of life of patients treated with chemotherapy. Future oncology (London, England). 2024:20(21):1515-1530. doi: 10.2217/fon-2023-0513. Epub 2024 Apr 8 [PubMed PMID: 38587388]
Level 2 (mid-level) evidenceChristmas NJ, Gordon CD, Murray TG, Tse D, Johnson T, Garonzik S, O'Brien JM. Intraorbital implants after enucleation and their complications: a 10-year review. Archives of ophthalmology (Chicago, Ill. : 1960). 1998 Sep:116(9):1199-203 [PubMed PMID: 9747679]
Ishtiaq R, Chaudhary MH, Rana MA, Jamil AR. Psychosocial implications of blindness and low vision in students of a school for children with blindness. Pakistan journal of medical sciences. 2016 Mar-Apr:32(2):431-4. doi: 10.12669/pjms.322.8737. Epub [PubMed PMID: 27182255]
Kaur K, Gurnani B. Low Vision Aids. StatPearls. 2024 Jan:(): [PubMed PMID: 36256771]
Kahán Z, Szántó I, Dudás R, Kapitány Z, Molnár M, Koncz Z, Mailáth M. Breast Cancer Survivorship Programme: Follow-Up, Rehabilitation, Psychosocial Oncology Care. 1st Central-Eastern European Professional Consensus Statement on Breast Cancer. Pathology oncology research : POR. 2022:28():1610391. doi: 10.3389/pore.2022.1610391. Epub 2022 Jun 2 [PubMed PMID: 35721327]
Level 3 (low-level) evidencevan Nispen RM, Virgili G, Hoeben M, Langelaan M, Klevering J, Keunen JE, van Rens GH. Low vision rehabilitation for better quality of life in visually impaired adults. The Cochrane database of systematic reviews. 2020 Jan 27:1(1):CD006543. doi: 10.1002/14651858.CD006543.pub2. Epub 2020 Jan 27 [PubMed PMID: 31985055]
Level 1 (high-level) evidenceFereidouni Z, Dehghan Abnavi S, Ghanbari Z, Gashmard R, Zarepour F, Khalili Samani N, Rajesh Sharma A, Ghasemi A. The Impact of Cancer on Mental Health and the Importance of Supportive Services. Galen medical journal. 2024:13():1-13. doi: 10.31661/gmj.v13i.3327. Epub 2024 Feb 26 [PubMed PMID: 39224547]
Da Costa GC, Aras MA, Chalakkal P, Da Costa MC. Ocular prosthesis incorporating IPS e-max press scleral veneer and a literature review on non-integrated ocular prosthesis. International journal of ophthalmology. 2017:10(1):148-156. doi: 10.18240/ijo.2017.01.24. Epub 2017 Jan 18 [PubMed PMID: 28149792]
Shao T, Verma HK, Pande B, Costanzo V, Ye W, Cai Y, Bhaskar LVKS. Physical Activity and Nutritional Influence on Immune Function: An Important Strategy to Improve Immunity and Health Status. Frontiers in physiology. 2021:12():751374. doi: 10.3389/fphys.2021.751374. Epub 2021 Oct 8 [PubMed PMID: 34690818]
Mahan M, Purt B. Ocular Trauma Prevention Strategies and Patient Counseling. StatPearls. 2025 Jan:(): [PubMed PMID: 35593844]
Devandan M, Daniel L, Sanjay AB. Prevalence of Ocular Injuries Following Road Traffic Accidents Among Two-Wheeler Riders/Pillions and the Factors Influencing Them Reported in a Tertiary Care Center in Chengalpattu in Tamil Nadu, India. Cureus. 2024 Oct:16(10):e72470. doi: 10.7759/cureus.72470. Epub 2024 Oct 27 [PubMed PMID: 39600737]
Ghassemi F, Khodabande A. Risk definition and management strategies in retinoblastoma: current perspectives. Clinical ophthalmology (Auckland, N.Z.). 2015:9():985-94. doi: 10.2147/OPTH.S59828. Epub 2015 Jun 8 [PubMed PMID: 26089630]
Level 3 (low-level) evidencePekacka A. The Role of Intraarterial Chemotherapy in the Management of Retinoblastoma. Journal of ophthalmology. 2020:2020():3638410. doi: 10.1155/2020/3638410. Epub 2020 Jan 24 [PubMed PMID: 32047660]
Mayer C, Gasalberti DP, Kumar A. Brachytherapy. StatPearls. 2025 Jan:(): [PubMed PMID: 32965861]
Manukonda R, Pujar A, Ramappa G, Vemuganti GK, Kaliki S. Identification of novel RB1 genetic variants in Retinoblastoma patients and their impact on clinical outcome. Ophthalmic genetics. 2022 Feb:43(1):64-72. doi: 10.1080/13816810.2021.1989602. Epub 2021 Oct 13 [PubMed PMID: 34645364]
Level 2 (mid-level) evidenceBrennan RC, Qaddoumi I, Billups CA, Free TL, Haik BG, Rodriguez-Galindo C, Wilson MW. Comparison of high-risk histopathological features in eyes with primary or secondary enucleation for retinoblastoma. The British journal of ophthalmology. 2015 Oct:99(10):1366-71. doi: 10.1136/bjophthalmol-2014-306364. Epub 2015 Apr 14 [PubMed PMID: 25873648]
de Vries ISA, van Ewijk R, Adriaansen LME, Bohte AE, Braat AJAT, Fajardo RD, Hiemcke-Jiwa LS, Hol MLF, Ter Horst SAJ, de Keizer B, Knops RRG, Meister MT, Schoot RA, Smeele LE, van Scheltinga ST, Vaarwerk B, Merks JHM, van Rijn RR. Imaging in rhabdomyosarcoma: a patient journey. Pediatric radiology. 2023 Apr:53(4):788-812. doi: 10.1007/s00247-023-05596-8. Epub 2023 Feb 27 [PubMed PMID: 36843091]
Sari NM, Hadiputri R, Kuntorini MS, Agustina H, Mardianty F. High-Risk Histopathologic Features of Retinoblastoma Treated at a Tertiary Hospital in West Java, Indonesia. Ocular oncology and pathology. 2021 Oct:7(5):353-360. doi: 10.1159/000517100. Epub 2021 Jul 20 [PubMed PMID: 34722492]
Tadic V, Ashcroft R, Brown JB, Dahrouge S. The Role of Social Workers in Interprofessional Primary Healthcare Teams. Healthcare policy = Politiques de sante. 2020 Aug:16(1):27-42. doi: 10.12927/hcpol.2020.26292. Epub [PubMed PMID: 32813638]
Hill JA, Gedleh A, Lee S, Hougham KA, Dimaras H. Knowledge, experiences and attitudes concerning genetics among retinoblastoma survivors and parents. European journal of human genetics : EJHG. 2018 Apr:26(4):505-517. doi: 10.1038/s41431-017-0027-9. Epub 2018 Jan 29 [PubMed PMID: 29379195]
V R, Chacko AM, Abdulla N, Annamalai M, Kandi V. Medication Adherence in Cancer Patients: A Comprehensive Review. Cureus. 2024 Jan:16(1):e52721. doi: 10.7759/cureus.52721. Epub 2024 Jan 22 [PubMed PMID: 38384629]
Demoor-Goldschmidt C, de Vathaire F. Review of risk factors of secondary cancers among cancer survivors. The British journal of radiology. 2019 Jan:92(1093):20180390. doi: 10.1259/bjr.20180390. Epub 2018 Sep 12 [PubMed PMID: 30102558]
van Dijk J, Grootenhuis MA, Imhof SM, Cohen-Kettenis PT, Moll AC, Huisman J. Coping strategies of retinoblastoma survivors in relation to behavioural problems. Psycho-oncology. 2009 Dec:18(12):1281-9. doi: 10.1002/pon.1507. Epub [PubMed PMID: 19222049]
Frazier TL, Lopez PM, Islam N, Wilson A, Earle K, Duliepre N, Zhong L, Bendik S, Drackett E, Manyindo N, Seidl L, Thorpe LE. Addressing Financial Barriers to Health Care Among People Who are Low-Income and Insured in New York City, 2014-2017. Journal of community health. 2023 Apr:48(2):353-366. doi: 10.1007/s10900-022-01173-6. Epub 2022 Dec 3 [PubMed PMID: 36462106]
Bello W, Hosotte C, Stampfli C, Pierrot A, Munier FL, Berger-Gryllaki M, Pezzatti J, Carrez L, Sadeghipour F. Stability study of topotecan in ophthalmic prefilled syringes for intravitreal delivery. European journal of hospital pharmacy : science and practice. 2025 Jan 20:():. pii: ejhpharm-2024-004346. doi: 10.1136/ejhpharm-2024-004346. Epub 2025 Jan 20 [PubMed PMID: 39837598]
Morrison-Jones V, West M. Post-Operative Care of the Cancer Patient: Emphasis on Functional Recovery, Rapid Rescue, and Survivorship. Current oncology (Toronto, Ont.). 2023 Sep 19:30(9):8575-8585. doi: 10.3390/curroncol30090622. Epub 2023 Sep 19 [PubMed PMID: 37754537]