Introduction
Turner syndrome, also referred to as congenital ovarian hypoplasia syndrome, was first described by Henri Turner, an Oklahoma physician in 1938.[1] It is the most common sex chromosomal abnormality found in females. It results when one of the X chromosomes is missing, partially or completely.
Etiology
Register For Free And Read The Full Article
 Search engine and full access to all medical articles
Search engine and full access to all medical articles- 10 free questions in your specialty
- Free CME/CE Activities
- Free daily question in your email
- Save favorite articles to your dashboard
- Emails offering discounts
Learn more about a Subscription to StatPearls Point-of-Care
Etiology
Turner syndrome results from a deletion or the non-functioning of one X chromosome in females. About half of the population with Turner syndrome have monosomy X (45,XO). The other 50% of the population has a mosaic chromosomal component (45,X with mosaicism).
Some types of anomalies in the X chromosome that can lead to a nonfunctioning X chromosome are as follows[2]:
- # Isochromosome Xq, where there are two copies of the long arm of the chromosome that are connected head to head.
- # Ring chromosome, where a part of the ends of short and long arms of the X chromosome is missing
- # Xp or Xq deletion, where the deletion of part of the short arm of the X chromosome takes place
Some patients with Turner syndrome can have a Y chromosome mosaicism. Although not a cause of Turner syndrome, the SHOX (short stature homeobox-containing gene on the X- chromosome) is associated with the short stature found in Turner syndrome. Turner syndrome is usually not inherited but is a random event during reproduction.
Epidemiology
As per the literature, Turner syndrome is seen in about 1 in 2000 to 1 in 2500 live female births.[3] However, the true prevalence remains unknown as many patients with a mild phenotype may remain undiagnosed or are diagnosed late in adulthood.[4] The occurrence of Turner syndrome is almost the same in different ethnicities and different countries. With increased awareness of prenatal ultrasound scans, the prevalence of Turner syndrome at birth is decreasing; this is because some mothers carrying fetuses with Turner syndrome choose to terminate the pregnancy.
Pathophysiology
Most instances of Turner syndrome are not inherited. When monosomy X is the cause, the chromosomal abnormality is a random event during the formation of reproductive cells in the person’s parent. An error in cell division is called nondisjunction and can result in reproductive cells with an abnormal number of chromosomes. For example, a sex chromosome can become lost from an egg or a sperm cell due to nondisjunction. If an atypical reproductive cell contributes to the genetic makeup of a child, each cell will possess a single X chromosome, and the other sex chromosome will be missing. See Image Turner Syndrome Karyotype.
Mosaic Turner syndrome is likewise not an inherited condition. It occurs due to a random event during the cell division stage in the early fetal development of the affected individual. As a result, some of a person's cells have the usual two sex chromosomes, while other cells contain only one copy of the X chromosome. Other sex chromosome abnormalities are possible in females with X chromosome mosaicism. Rarely, Turner syndrome can result from a partial deletion of the X chromosome, and this can pass from one generation to the next.
History and Physical
Turner syndrome can be identified prenatally with abnormal ultrasound findings of increased nuchal translucency, nuchal cystic hygroma, coarctation of the aorta/left-sided cardiac anomalies, brachycephaly, horseshoe kidney, polyhydramnios, oligohydramnios or non-immune fetal hydrops. In the female newborn, Turner syndrome can present with congenital lymphedema of the hands and feet, webbed neck, nail dysplasia, narrow and high-arched palate, and short fourth metacarpals or metatarsals. As they grow up, the girls develop short stature, “shield” chest with widely spaced nipples, webbed neck, low hairline at the base of the neck, cubitus valgus, and Madelung deformity of the forearm and the wrist.[5]
Patients with Turner syndrome usually have normal intelligence but may have specific neurocognitive deficits, e.g., problems with visuospatial organization. This situation can lead to an increased risk of learning disabilities, especially involving calculations, memory, and attention. In adolescence, females will often present with delayed puberty or primary amenorrhea, secondary to premature ovarian failure. “Streak gonads” are a characteristic of Turner syndrome. These are the ovaries, mainly consisting of connective tissue and no follicles or only a few atretic follicles.
Patients with Turner syndrome also have an increased risk of cardiovascular malformations, which in turn leads to increased mortality risk in these individuals. Some of the cardiac malformations are aortic valve abnormalities (primarily bicuspid aortic valve), elongated transverse aortic arch, pulmonary venous anomalies. Aortic dissection further increases the risk of death in these patients. Hearing loss is common due to either recurrent otitis media causing conductive hearing loss, or due to the defect in the outer hair cells on the cochlea causing sensorineural hearing loss [6]. Renal anomalies are common in Turner syndrome and include collecting system malformations, positional abnormalities, and horseshoe kidneys.
Ocular abnormalities can present with Turner syndrome, such as nearsightedness or farsightedness, strabismus, amblyopia, epicanthic folds, ptosis, hypertelorism, and red-green color blindness.[7] Turner syndrome increases the risk of autoimmune disorders, including hypothyroidism, celiac disease, and inflammatory bowel disease.[8] Due to the presence of dysgenic gonads, females with Turner syndrome are at an increased risk of developing gonadoblastoma.
Evaluation
Turner syndrome may be prenatally diagnosed by chorionic-villus sampling or amniocentesis. Turner syndrome should be suspected when a prenatal ultrasound shows fetal hydrops, cystic hygroma, or cardiac defects. The diagnosis requires confirmation after birth with karyotype testing. Occasionally, the karyotype can be normal if it is mosaicism, and if there is a strong suspicion, a FISH study is an option in addition to the karyotype. Genetic testing with karyotype analysis is necessary to confirm the diagnosis in individuals with characteristic clinical features described above. The first step is a karyotype analysis with peripheral blood mononuclear cells.
In adolescence, the patients can present with either delayed onset of puberty or amenorrhea. Elevated levels of follicle-stimulating hormone (FSH) are suggestive of Turner syndrome, and the anti-Mullerian hormone (AMH) may be a more sensitive marker for predicting ovarian failure.[9] If the initial karyotype is normal in a patient with clinically suspected Turner syndrome, a second karyotype should be performed using a different tissue like skin, buccal mucosa cells, or bladder epithelial cells.
Following a diagnosis of Turner syndrome, management includes evaluation for other associated abnormalities like cardiac anomalies, renal anomalies, and learning disabilities. Screening should be a part of the baseline evaluation, and patients should undergo periodic screening thereafter. At initial diagnosis, patients should get renal ultrasonography and cardiovascular evaluation, including echocardiography in infants and children, and MRI in older girls and women.[10][11][12]
Some of the screening laboratory tests include:
At four years of age and above: serum TSH to screen for autoimmune thyroiditis and tissue transglutaminase with total IgA to screen for celiac disease.
At ten years of age and above: fasting blood glucose, glycated hemoglobin, ALT, AST, serum creatinine, and urinalysis to screen for diabetes mellitus, fatty liver, and kidney disease.
Treatment / Management
Short Stature
- Girls with Turner syndrome generally have short stature, and their growth requires close monitoring. Turner syndrome does not cause growth hormone deficiency. But, the patients respond well to growth hormone therapy and should be started on treatment with growth hormone once their height falls below 5% for age. If not treated appropriately, the estimates are that the adult height would be 20 cm below the average adult female height. Growth hormone therapy should continue until the patients reach their adult height and no longer have any growth potential. Once the patient is on the growth hormone therapy, it can sometimes expose underlying scoliosis. Hence, the patients should have their spines monitored closely during the treatment. If patients develop scoliosis, then they should be referred to as orthopedic surgery for possible bracing or corrective surgery. Some other possible adverse effects of the growth hormone treatment are intracranial hypertension, slipped capital femoral epiphyses, and pancreatitis. If the patient requires further assistance for growth in addition to growth hormone, oxandrolone, or delayed, pubertal induction can be offered.
Cardiac
- Cardiac abnormalities are frequently associated with Turner syndrome. At the time of diagnosis, the patient should be evaluated by a cardiologist with an EKG to evaluate for prolonged QT interval. Blood pressure should be measured in the upper and lower extremities, and patients should get an echocardiogram or cardiac MRI to look for cardiac anomalies.[13]
- For prolonged QT interval, QT-prolonging drugs (antiarrhythmics, macrolide and fluoroquinolones, metronidazole, some antifungals, and antiretrovirals, psychiatric medications) should be avoided. If coarctation of the aorta is present, it requires corrective surgery. Throughout life, the patients require monitoring for aortic dilation with echocardiograms or cardiac MRI. Blood pressure should be maintained within the normal range to help decrease the risk of aortic dilation and dissection. Blood pressure should be controlled using beta-blockers as first-line treatment, followed by an ACE inhibitor.[13]
Cognitive function/learning disabilities
- Girls with Turner syndrome often have learning disabilities despite normal intelligence, which might warrant special education and assessments at school.
Hearing loss
- Regular monitoring of hearing, including serial audiology evaluations, is recommended throughout life, with an audiology evaluation every 3 yrs in children and every five years in adults.
Renal
- A renal ultrasound is necessary at the time of diagnosis. Renal abnormalities frequently present with Turner syndrome, including collecting system malformations, positional/horseshoe kidney, and mal-rotated kidneys. Obstruction due to ureteropelvic junction abnormalities can cause hydronephrosis and increase the risk of pyelonephritis.[12] If any abnormalities are present, the patient should receive a referral to nephrology.
Ovarian Failure
- Girls with Turner syndrome usually present with primary amenorrhea or delayed puberty secondary to premature ovarian failure. Serum FSH and AMH should be measured at around 10 to 11 years of age. Serum AMH can help predict ovarian function, and patients with detectable levels are likely to experience spontaneous puberty.[14] Estrogen replacement therapy should start if no breast development begins at around 11 to 12 years of age.
- Estrogen therapy - Almost all girls with Turner syndrome need estrogen, even if they have spontaneous puberty, which may persist for some time but is usually followed by primary ovarian insufficiency. Later, cyclic progestins are added to the regimen to induce cyclic uterine bleeding and prevent endometrial hyperplasia. Estrogen therapy should be initiated at around 11 to 12 years of age if gonadotropins are elevated or AMH level is low. The treatment can begin at doses between 1/10 to 1/8 of adult replacement dose and gradually increased every six months to simulate normal pubertal progression until reaching adult dosing.
- Turner syndrome patients will often have the option for cryopreservation of ovarian tissue or oocytes. This option is only available to those with evidence of ovarian function and is not recommended prior to 12 years of age.
- Due to ovarian follicular depletion, most women with Turner syndrome are infertile. In vitro fertilization with a donor, oocytes is an option to conceive.
Osteoporosis/Bone Health
- Patients with Turner syndrome have an increased risk of low bone mineral density and fractures. Their risk becomes lowered with estrogen therapy and supplemental vitamin D and calcium.[15] Turner syndrome also increases the risk for scoliosis, and patients should have screening annually and every six months when receiving growth hormone therapy.
Screening for other comorbidities
- Celiac disease – Tissue transglutaminase immunoglobulin A antibodies should be measured at around two years of age and repeated every two years throughout childhood.[13]
- Autoimmune thyroiditis – TSH and free or total T4 should be measured annually, beginning around four years.
- Liver disease – ALT, AST, GGT, and alkaline phosphatase should be measured annually after ten years of age. These labs are usually elevated in Turner syndrome, but if persistent and greater than twice normal, it requires further evaluation by a hepatologist.
- Metabolic syndrome – Hemoglobin A1c should be measured annually, beginning at ten years of age to screen for hyperglycemia. Patients should also have screening for dyslipidemia by measuring a lipid panel annually if at least one cardiovascular disease risk factor is present.
- Vit D deficiency – Serum 25-hydroxyvitamin D should be measured between 9 to11 years of age and every 2 to 3 years thereafter.
- Gonadoblastoma – Patients with Turner syndrome who have marker chromosome elements on karyotype or patients who develop virilization, should be screened for the Y chromosome. If the Y chromosome is present, gonads should be removed; otherwise, it increases the risk of gonadoblastoma.[16] (B2)
Differential Diagnosis
Noonan syndrome is a condition that is very similar to Turner syndrome and often can be confusing to distinguish. Noonan syndrome presents with similar clinical characteristics such as a webbed neck, short stature, cardiac, and renal abnormalities. In Noonan syndrome, there is no chromosomal abnormality, unlike in Turner syndrome with the absent or non-functioning X chromosome. Therefore, Noonan syndrome can be seen in both males and females, whereas Turner syndrome is seen only in females. Genetic testing is required to differentiate between these two conditions.[17]
Prognosis
Patients with Turner syndrome have an increased mortality rate, three times greater than the general population. Cardiovascular disease due to coronary heart disease and stroke in older patients is a significant factor. Of the congenital cardiovascular disease, an aortic aneurysm is the largest cause. Patients also demonstrate an increase in mortality due to pneumonia, diabetes, epilepsy, liver disease, and kidney disease.[16]
Complications
Some common complications or associated findings with Turner syndrome are:
- Hearing loss
- Hypothyroidism
- Liver function abnormalities
- Neurocognitive deficits and may require special services in school.
- Increased risk of autoimmune diseases
- Aortic dissection
- Gonadoblastoma risk increases in the women with the Y chromosome
- Metabolic disorders – central obesity, insulin resistance, diabetes mellitus type 2, dyslipidemia
Consultations
- Endocrinology
- Cardiology
- Gynecology
- Audiology
- Ophthalmology
- Orthopedic surgery (if they develop scoliosis)
- Nephrology (if they have renal abnormalities)
Deterrence and Patient Education
Turner syndrome is a lifelong diagnosis and requires consistent follow-up. Many complications are associated with the disease, such as short stature, heart and kidney malformations, ovarian failure, increased risk for obesity, diabetes, and elevated blood pressure. A lot of these problems will require multiple specialists to be involved in care.
Pearls and Other Issues
- A karyotype is necessary to diagnose Turner syndrome.
- There can be many problems associated with Turner syndrome requiring multiple specialists to be involved. The difficulties include short stature, heart, and renal abnormalities, hearing loss, scoliosis or other orthopedic issues, eye abnormalities, metabolic syndrome, and increased risk for autoimmune disease.
- These patients require frequent screening throughout life to monitor for all the possible complications.
Enhancing Healthcare Team Outcomes
Patients with Turner syndrome have a lot of comorbidities and require many specialty fields to be involved in care, functioning as an interprofessional team. Although the primary provider should be following and managing many of the screening and follow up care, the other specialties involved should include cardiology, endocrinology, obstetrics and gynecology, ophthalmology, audiology, nephrology, and orthopedic surgery. It is essential for the different specialists to communicate well with each other and especially the primary team, so that they can address all the potential complications. Nurses who have specialized training in genetics provide patient education, monitor patients, and coordinate referrals and communication between team members. Pharmacists review hormonal therapies and work with patients and their families about administration and compliance. The interprofessional team approach will lead to optimal outcomes for Turner syndrome patients. [Level 5]
Media
(Click Image to Enlarge)

Girl With Turner Syndrome. Photos showing before and immediately after her operation for neck-webbing.
Contributed by Wikimedia Commons (CC BY 2.0) https://creativecommons.org/licenses/by/2.0/deed.en
(Click Image to Enlarge)
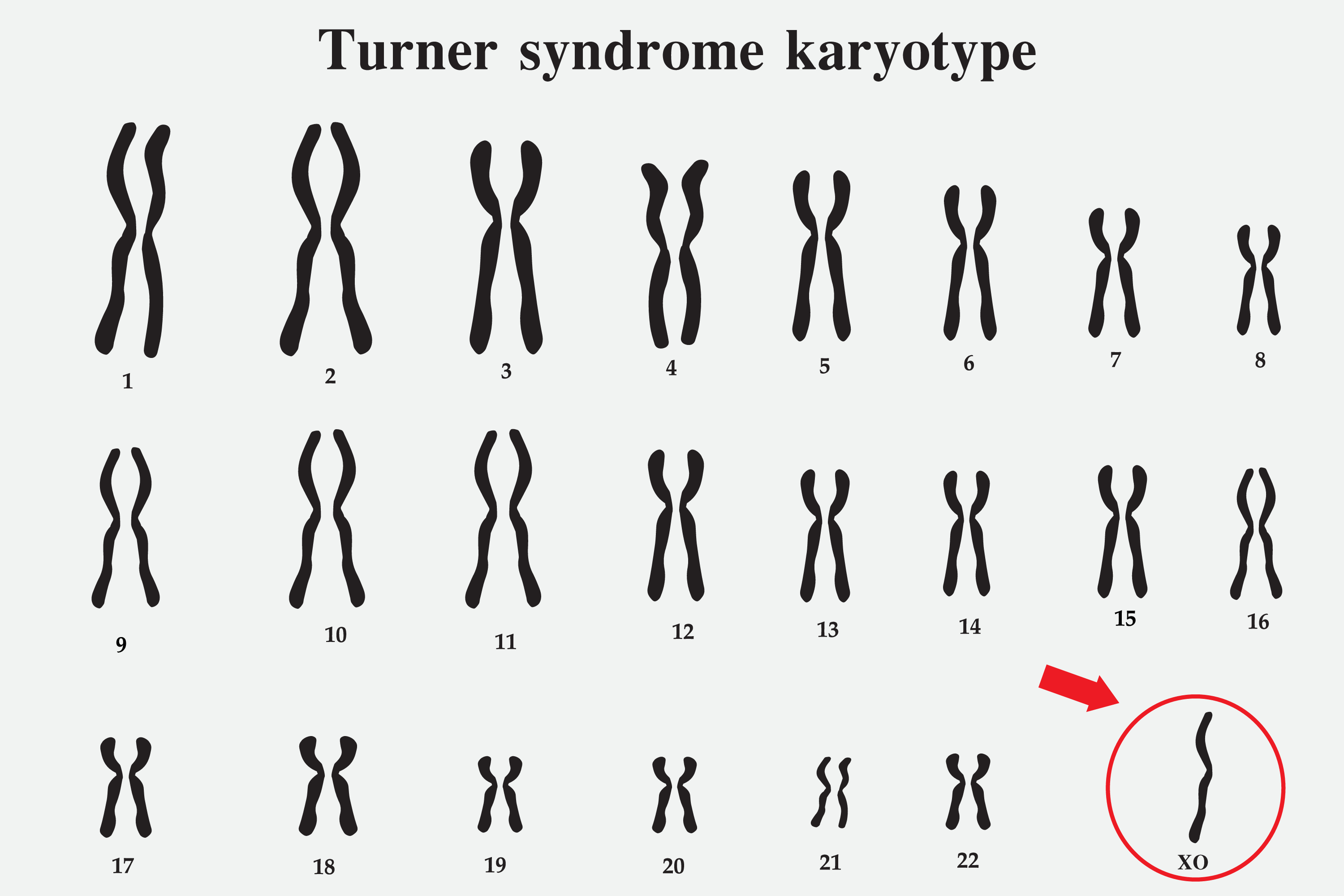
Turner Syndrome Karyotype
Shutterstock. 1455406670
References
. Classic pages in obstetrics and gynecology by Henry H. Turner. A syndrome of infantilism, congenital webbed neck, and cubitus valgus. Endocrinology, vol. 23, pp. 566-574, 1938. American journal of obstetrics and gynecology. 1972 May 15:113(2):279 [PubMed PMID: 4557013]
Wolff DJ, Van Dyke DL, Powell CM, Working Group of the ACMG Laboratory Quality Assurance Committee. Laboratory guideline for Turner syndrome. Genetics in medicine : official journal of the American College of Medical Genetics. 2010 Jan:12(1):52-5. doi: 10.1097/GIM.0b013e3181c684b2. Epub [PubMed PMID: 20081420]
Level 2 (mid-level) evidenceCui X, Cui Y, Shi L, Luan J, Zhou X, Han J. A basic understanding of Turner syndrome: Incidence, complications, diagnosis, and treatment. Intractable & rare diseases research. 2018 Nov:7(4):223-228. doi: 10.5582/irdr.2017.01056. Epub [PubMed PMID: 30560013]
Level 3 (low-level) evidenceGunther DF, Eugster E, Zagar AJ, Bryant CG, Davenport ML, Quigley CA. Ascertainment bias in Turner syndrome: new insights from girls who were diagnosed incidentally in prenatal life. Pediatrics. 2004 Sep:114(3):640-4 [PubMed PMID: 15342833]
Sävendahl L, Davenport ML. Delayed diagnoses of Turner's syndrome: proposed guidelines for change. The Journal of pediatrics. 2000 Oct:137(4):455-9 [PubMed PMID: 11035820]
Barrenäs ML, Nylén O, Hanson C. The influence of karyotype on the auricle, otitis media and hearing in Turner syndrome. Hearing research. 1999 Dec:138(1-2):163-70 [PubMed PMID: 10575123]
Level 1 (high-level) evidenceAdhikary HP. Ocular manifestations of Turner's syndrome. Transactions of the ophthalmological societies of the United Kingdom. 1981:101 (Pt 4)():395-6 [PubMed PMID: 6964261]
Lowenstein EJ, Kim KH, Glick SA. Turner's syndrome in dermatology. Journal of the American Academy of Dermatology. 2004 May:50(5):767-76 [PubMed PMID: 15097963]
Lunding SA, Aksglaede L, Anderson RA, Main KM, Juul A, Hagen CP, Pedersen AT. AMH as Predictor of Premature Ovarian Insufficiency: A Longitudinal Study of 120 Turner Syndrome Patients. The Journal of clinical endocrinology and metabolism. 2015 Jul:100(7):E1030-8. doi: 10.1210/jc.2015-1621. Epub 2015 May 15 [PubMed PMID: 25978111]
Kim HK, Gottliebson W, Hor K, Backeljauw P, Gutmark-Little I, Salisbury SR, Racadio JM, Helton-Skally K, Fleck R. Cardiovascular anomalies in Turner syndrome: spectrum, prevalence, and cardiac MRI findings in a pediatric and young adult population. AJR. American journal of roentgenology. 2011 Feb:196(2):454-60. doi: 10.2214/AJR.10.4973. Epub [PubMed PMID: 21257900]
Level 2 (mid-level) evidenceMortensen KH, Young L, De Backer J, Silberbach M, Collins RT, Duijnhouwer AL, Pandya B, Gravholt CH, Lopez L, Roos-Hesselink JW. Cardiovascular imaging in Turner syndrome: state-of-the-art practice across the lifespan. Heart (British Cardiac Society). 2018 Nov:104(22):1823-1831. doi: 10.1136/heartjnl-2017-312658. Epub 2018 Sep 18 [PubMed PMID: 30228249]
Bilge I, Kayserili H, Emre S, Nayir A, Sirin A, Tukel T, Bas F, Kilic G, Basaran S, Gunoz H, Apak M. Frequency of renal malformations in Turner syndrome: analysis of 82 Turkish children. Pediatric nephrology (Berlin, Germany). 2000 Oct:14(12):1111-4 [PubMed PMID: 11045397]
Bondy CA, Turner Syndrome Study Group. Care of girls and women with Turner syndrome: a guideline of the Turner Syndrome Study Group. The Journal of clinical endocrinology and metabolism. 2007 Jan:92(1):10-25 [PubMed PMID: 17047017]
Klein KO, Rosenfield RL, Santen RJ, Gawlik AM, Backeljauw PF, Gravholt CH, Sas TCJ, Mauras N. Estrogen Replacement in Turner Syndrome: Literature Review and Practical Considerations. The Journal of clinical endocrinology and metabolism. 2018 May 1:103(5):1790-1803. doi: 10.1210/jc.2017-02183. Epub [PubMed PMID: 29438552]
Bakalov VK, Chen ML, Baron J, Hanton LB, Reynolds JC, Stratakis CA, Axelrod LE, Bondy CA. Bone mineral density and fractures in Turner syndrome. The American journal of medicine. 2003 Sep:115(4):259-64 [PubMed PMID: 12967689]
Schoemaker MJ, Swerdlow AJ, Higgins CD, Wright AF, Jacobs PA, United Kingdom Clinical Cytogenetics Group. Mortality in women with turner syndrome in Great Britain: a national cohort study. The Journal of clinical endocrinology and metabolism. 2008 Dec:93(12):4735-42. doi: 10.1210/jc.2008-1049. Epub 2008 Sep 23 [PubMed PMID: 18812477]
Level 2 (mid-level) evidenceRoberts AE, Allanson JE, Tartaglia M, Gelb BD. Noonan syndrome. Lancet (London, England). 2013 Jan 26:381(9863):333-42. doi: 10.1016/S0140-6736(12)61023-X. Epub 2013 Jan 10 [PubMed PMID: 23312968]